دوره NGS منتشر شد …

Biopython (بایوپایتون) یک مجموعه ابزار نرمافزاری متنباز و رایگان است که به محققان حوزه بیوانفورماتیک امکان میدهد تا تحلیلهای پیچیده ژنتیکی و پروتئینی را در محیط برنامهنویسی پایتون انجام دهند. این کتابخانه در سال 1999 با هدف تسهیل تحقیقات زیستشناسی محاسباتی آغاز به کار کرد و اکنون به یکی از اصلیترین ابزارهای مورداستفاده در بیوانفورماتیک تبدیل شده است. Biopython با ارائه مجموعهای از توابع و کلاسهای آماده، به محققان کمک میکند تا بهجای صرف زمان برای برنامهنویسی الگوریتمهای پایه، بر اهداف علمی خود تمرکز کنند. از مزایای اصلی Biopython میتوان به سادگی استفاده، انعطافپذیری، جامعه فعال توسعهدهندگان و مستندات گسترده آن اشاره کرد.

پیش از نصب Biopython، لازم است محیط پایتون مناسبی روی سیستم خود داشته باشید. توصیه میشود از پایتون نسخه 3.6 یا بالاتر استفاده کنید. همچنین، برخی از ماژولهای Biopython (بایوپایتون) به کتابخانههای خارجی مانند NumPy وابستهاند که برای تحلیلهای عددی و محاسبات ماتریسی کاربرد دارند. برای تجربه بهتر، استفاده از محیطهای مجازی مانند Anaconda یا virtualenv پیشنهاد میشود که امکان مدیریت وابستگیها را به شکل جداگانه فراهم میکنند. این رویکرد به خصوص برای پروژههای بزرگ یا هنگام کار با نسخههای مختلف کتابخانهها مفید است.
سادهترین روش نصب Biopython، کار با pip بهعنوان package manager پایتون است:
pip install biopythonبرای کاربران Anaconda، میتوانید از دستور زیر استفاده کنید:
conda install -c conda-forge biopythonپس از نصب موفق، میتوانید با وارد کردن دستور زیر در محیط پایتون، از نصب صحیح آن اطمینان حاصل کنید:
import Bio
print(Bio.__version__)دیدن نسخه Biopython، بهمعنی نصب موفقیتآمیز آن است. برای کاربران پیشرفتهتر، امکان نصب از کد منبع نیز وجود دارد که انعطافپذیری بیشتری برای سفارشیسازی فراهم میکند. لینک سایت رسمی بایوپایتون.
در هسته Biopython، کلاس Seq قرار دارد که برای نمایش و کار با توالیهای زیستی مانند DNA، RNA و پروتئین طراحی شده است. این کلاس امکان انجام عملیات متداول مانند ترجمه DNA به پروتئین، معکوسسازی و مکملسازی توالیها را فراهم میکند. علاوه بر Seq، کلاس SeqRecord برای نگهداری اطلاعات اضافی مرتبط با توالیها مانند شناسه، توضیحات و ویژگیهای دیگر استفاده میشود. این ساختار داده، امکان کار با فایلهای استاندارد زیستی را بهشکلی سادهتر فراهم میسازد و باعث میشود انتقال دیتا بین فرمتهای مختلف بدون از دست دادن اطلاعات مهم انجام شود.
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
record = SeqRecord(
Seq("MKQHKAMIVALIVICITAVVAALVTRKDLCEVHIRTGQTEVAVF"),
id="YP_025292.1",
name="HokC",
description="toxic membrane protein, small",
)
print(record)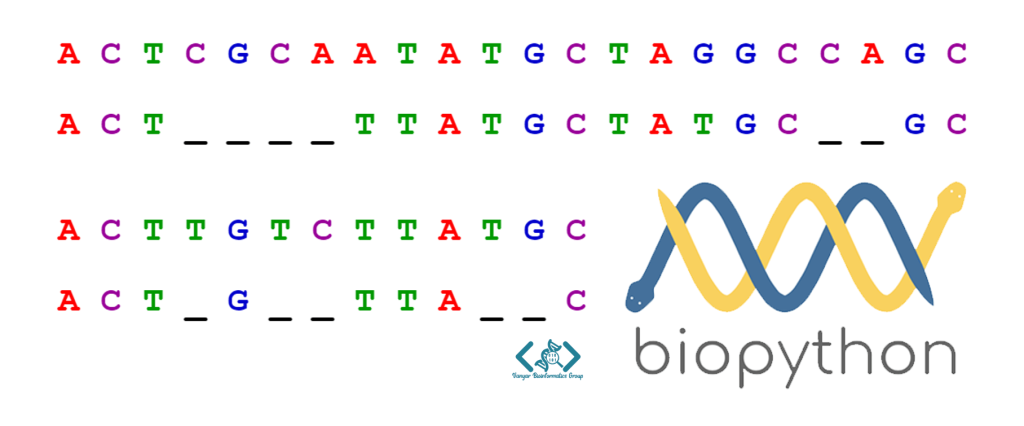
Biopython توانایی قابل توجهی در پردازش فایلهای با فرمتهای مختلف زیستی دارد. از پرکاربردترین این فرمتها میتوان به FASTA (برای توالیهای ساده)، GenBank (شامل توالی و اطلاعات تفصیلی)، EMBL، Swiss-Prot و PDB (برای ساختارهای سهبعدی پروتئین) اشاره کرد. ماژول Bio.SeqIO در Biopython، یک رابط متحدالشکل برای خواندن و نوشتن این فرمتهای متنوع ارائه میدهد. این ماژول با انتزاع جزئیات فنی هر فرمت، به محققان اجازه میدهد روی تحلیل دادهها تمرکز کنند، بدون اینکه نگران پیچیدگیهای ساختاری فایلها باشند.
یکی از قابلیتهای مهم Biopython، امکان دسترسی به دیتابیسهای آنلاین از درون کد پایتون است. ماژول Bio.Entrez امکان ارتباط با سیستم Entrez NCBI را فراهم میکند که دربرگیرنده پایگاههای مهمی مانند GenBank، PubMed و OMIM است. با استفاده از این ماژول، محققان میتوانند جستجو، دریافت و تحلیل دادههای زیستی را بهصورت برنامهنویسیشده انجام دهند. این قابلیت بهخصوص برای تحقیقات گسترده که نیازمند استخراج حجم زیادی از دیتا است، ارزشمند میباشد.
from Bio import Entrez
Entrez.email = "Your.Name.Here@example.org"
handle = Entrez.einfo() # or esearch, efetch, ...
record = Entrez.read(handle)
handle.close()handle = Entrez.esummary(db="pubmed", id="19304878,14630660", retmode="xml")
records = Entrez.parse(handle)
for record in records:
# each record is a Python dictionary or list.
print(record['Title'])
Biopython: freely available Python tools for computational molecular biology and bioinformatics.
PDB file parser and structure class implemented in Python.
handle.close()ماژول Bio.PDB در Biopython ابزاری قدرتمند برای کار با ساختارهای سهبعدی پروتئینها ارائه میدهد. این ماژول امکان دریافت فایلهای PDB، تجزیه و تحلیل آنها و استخراج اطلاعات مهمی مانند موقعیت فضایی اتمها، زنجیرههای پلیپپتیدی و مدلهای مختلف را فراهم میسازد. با استفاده از Bio.PDB، محققان میتوانند محاسبات هندسی مانند فاصله بین اتمها، زوایای پیوندی و تحلیلهای پیچیدهتر را انجام دهند. این قابلیت برای مطالعات ساختاری پروتئین، مدلسازی مولکولی و طراحی دارو بسیار کاربردی است.
from Bio.PDB import PDBParser
from Bio.PDB.DSSP import DSSP
p = PDBParser()
structure = p.get_structure("1MOT", "/local-pdb/1mot.pdb")
model = structure[0]
dssp = DSSP(model, "/local-pdb/1mot.pdb")همردیفسازی توالیها یکی از اساسیترین عملیات در بیوانفورماتیک است که برای مقایسه توالیها و شناسایی مناطق مشابه استفاده میشود. Biopython امکان انجام همردیفسازیهای دو توالی (pairwise alignment) و همردیفسازیهای چندگانه (multiple sequence alignment) را با استفاده از الگوریتمهای متفاوت فراهم میکند. ماژولهای Bio.pairwise2 و Bio.Align امکان همردیفسازی محلی و سراسری را ارائه میدهند. همچنین، Biopython میتواند به عنوان یک واسط برای ابزارهای خارجی معروف مانند BLAST، Clustal و MUSCLE عمل کند. این قابلیت به محققان امکان میدهد تا بدون خروج از محیط برنامهنویسی پایتون، از الگوریتمهای قدرتمند و بهینه استفاده کنند.
Bio.pairwise2:
from Bio import pairwise2
alignments = pairwise2.align.globalxx("ACCGT", "ACG")from Bio.pairwise2 import format_alignment
print(format_alignment(*alignments[0]))
ACCGT
| ||
A-CG-
Score=3Bio.Align:
from Bio import AlignIO
align = AlignIO.read("Clustalw/opuntia.aln", "clustal")
print(align)
SingleLetterAlphabet() alignment with 7 rows and 156 columns
TATACATTAAAGAAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA gi|6273285|gb|AF191659.1|AF191
TATACATTAAAGAAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA gi|6273284|gb|AF191658.1|AF191
TATACATTAAAGAAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA gi|6273287|gb|AF191661.1|AF191
TATACATAAAAGAAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA gi|6273286|gb|AF191660.1|AF191
TATACATTAAAGGAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA gi|6273290|gb|AF191664.1|AF191
TATACATTAAAGGAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA gi|6273289|gb|AF191663.1|AF191
TATACATTAAAGGAGGGGGATGCGGATAAATGGAAAGGCGAAAG...AGA gi|6273291|gb|AF191665.1|AF191Biopython ابزارهای متنوعی برای تحلیل ویژگیهای فیزیکوشیمیایی توالیهای زیستی ارائه میدهد. ماژول Bio.SeqUtils امکان محاسبه پارامترهایی مانند ترکیب نوکلئوتیدی، درصد GC، نقطه ایزوالکتریک (pI) پروتئینها و وزن مولکولی را فراهم میسازد. این اطلاعات در مطالعات مختلف از جمله طراحی پرایمر، بررسی پایداری پروتئین و مطالعات تکاملی کاربرد دارند. علاوه بر این، ماژولهای تخصصی مانند Bio.SeqUtils.ProtParam امکان محاسبه شاخصهای پایداری، آبگریزی و دیگر پارامترهای مهم پروتئینی را فراهم میکنند.
from Bio.SeqUtils.ProtParam import ProteinAnalysis
X = ProteinAnalysis("MAEGEITTFTALTEKFNLPPGNYKKPKLLYCSNGGHFLRILPDGTVDGT"
"RDRSDQHIQLQLSAESVGEVYIKSTETGQYLAMDTSGLLYGSQTPSEEC"
"LFLERLEENHYNTYTSKKHAEKNWFVGLKKNGSCKRGPRTHYGQKAILF"
"LPLPV")
print(X.count_amino_acids()['A'])
6
print(X.count_amino_acids()['E'])
12
print("%0.2f" % X.get_amino_acids_percent()['A'])
0.04
print("%0.2f" % X.get_amino_acids_percent()['L'])
0.12
print("%0.2f" % X.molecular_weight())
17103.16
print("%0.2f" % X.aromaticity())
0.10
print("%0.2f" % X.instability_index())
41.98
print("%0.2f" % X.isoelectric_point())
7.72
sec_struc = X.secondary_structure_fraction() # [helix, turn, sheet]
print("%0.2f" % sec_struc[0]) # helix
0.28
epsilon_prot = X.molar_extinction_coefficient() # [reduced, oxidized]
print(epsilon_prot[0]) # with reduced cysteines
17420
print(epsilon_prot[1]) # with disulfid bridges
17545
آنالیز فیلوژنتیک روشی قدرتمند برای بررسی روابط تکاملی میان موجودات زنده با استفاده از دادههای مولکولی است. Biopython از طریق ماژول Bio.Phylo امکان کار با درختهای فیلوژنتیک را فراهم میسازد. با استفاده از این ماژول، میتوانید درختها را از فرمتهای استاندارد مانند Newick، NEXUS و PhyloXML بخوانید و تحلیل کنید. همچنین امکان نمایش گرافیکی درختها با استفاده از کتابخانههایی مانند Matplotlib وجود دارد. ماژول Bio.Phylo تحلیلهای متنوعی مانند محاسبه فاصله بین گونهها، شناسایی گرههای مشترک و عملیات پیمایش درخت را امکانپذیر میسازد. این قابلیتها برای متخصصان زیستشناسی تکاملی، تاکسونومی و اپیدمیولوژی مولکولی بسیار ارزشمند است.
>>> tree = Phylo.parse('phyloxml_examples.xml', 'phyloxml').next()
>>> print(tree)
Phylogeny(description='phyloXML allows to use either a "branch_length"
attribute or element to indicate branch lengths.', name='example from
Prof. Joe Felsenstein s book "Inferring Phylogenies"')
Clade()
Clade(branch_length=0.06)
Clade(branch_length=0.102, name='A')
Clade(branch_length=0.23, name='B')
Clade(branch_length=0.4, name='C')
...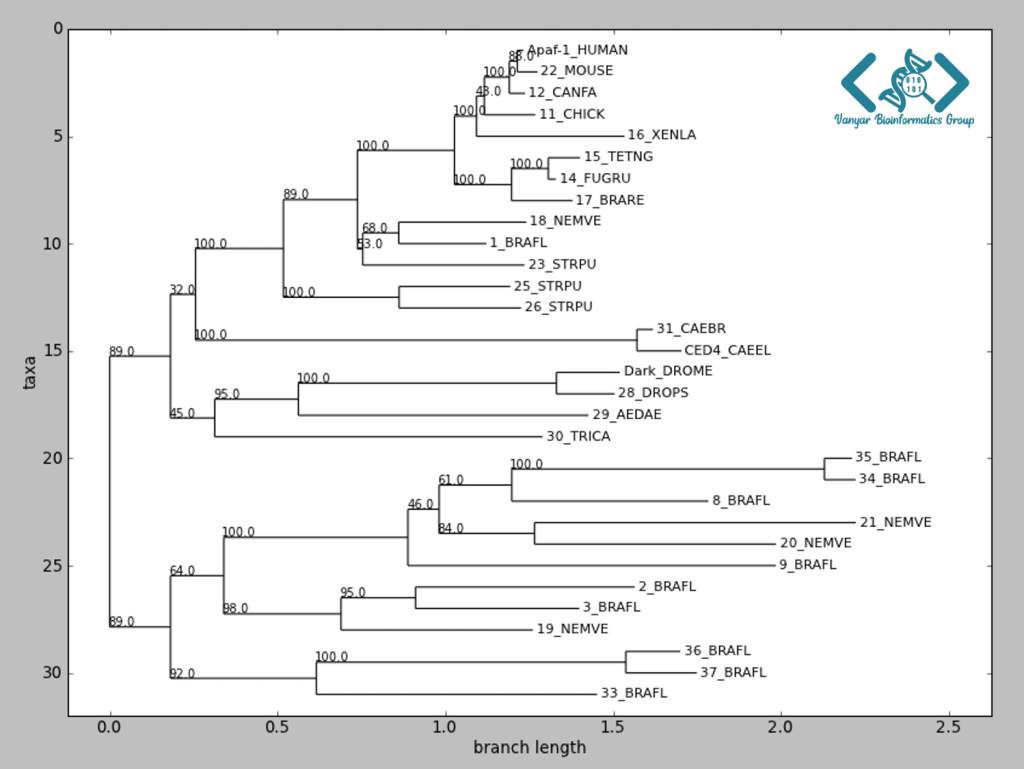
محاسبه فواصل تکاملی بین توالیها یکی از گامهای مهم در آنالیزهای فیلوژنتیک است. Biopython امکان محاسبه این فواصل را با استفاده از مدلهای مختلف تکاملی فراهم میکند. پس از محاسبه فواصل، میتوان از الگوریتمهای مختلف برای ساخت درخت استفاده کرد. یکی از مزایای استفاده از بایوپایتون، امکان ترکیب آن با کتابخانههای قدرتمند ترسیم مانند Matplotlib است که به محققان امکان میدهد درختهای فیلوژنتیک را به شکلهای مختلف و با جزئیات سفارشی نمایش دهند. این ویژگی به خصوص برای انتشار نتایج و ارائه یافتههای تحقیقاتی مفید است.
یکی از نقاط قوت Biopython، توانایی آن در برقراری ارتباط با نرمافزارها و ابزارهای خارجی است. ماژولهای متعددی مانند Bio.Blast، Bio.Clustalw و Bio.Emboss به عنوان واسط برای ابزارهای محبوب بیوانفورماتیک عمل میکنند. این قابلیت به محققان امکان میدهد فرآیند استفاده از این ابزارها را از طریق اسکریپتهای پایتون خودکارسازی کنند. همچنین، امکان پردازش و تحلیل نتایج حاصل از این ابزارها به صورت یکپارچه وجود دارد. این ویژگی به خصوص برای ایجاد pipelines تحلیلی که از چندین ابزار مختلف استفاده میکنند، بسیار مفید است و باعث افزایش بهرهوری و کاهش خطاهای کاربر میشود.
با گسترش تکنیکهای توالییابی نسل بعدی، حجم دادههای ژنومی بهطور چشمگیری افزایش یافته است. اگرچه Biopython در ابتدا برای کار با حجم کمتری از دیتا طراحی شده بود، اما با افزودن ماژولهای جدید و بهینهسازیهای مختلف، امکان پردازش دادههای NGS را نیز فراهم کرده است. به عنوان مثال، ماژول Bio.SeqIO.QualityIO امکان کار با فایلهای FASTQ را که فرمت استاندارد برای دادههای NGS هستند، فراهم میسازد. همچنین، با ترکیب بایوپایتون و کتابخانههای کارآمد دیگر مانند NumPy و pandas، میتوان پردازش حجم زیادی از دادههای ژنومی را به صورت کارآمد انجام داد. این قابلیت برای تحلیلهای RNA-Seq، ChIP-Seq و متیلاسیون DNA بسیار کاربردی است.
Biopython یک پروژه متنباز فعال است که به طور مداوم توسط جامعه جهانی توسعهدهندگان در حال بهبود و گسترش است. نسخههای جدید این کتابخانه معمولاً هر چند ماه یک بار منتشر میشوند که شامل رفع باگها، بهبود کارایی و افزودن قابلیتهای جدید هستند. در سالهای اخیر، تمرکز توسعه Biopython بر سازگاری با پایتون 3، بهبود عملکرد در پردازش دادههای حجیم و افزودن پشتیبانی از فرمتهای جدید دادههای زیستی بوده است.
توسعهدهندگان بایوپایتون همواره در جهت بهبود مستندات و ارائه مثالهای عملی بیشتر، تلاش کردهاند. پیگیری اخبار و بهروزرسانیهای بایوپایتون از طریق وبسایت رسمی، لیست ایمیل و مخزن GitHub آن امکانپذیر است.
یکی از نقاط قوت پروژههای متنباز مانند Biopython، امکان مشارکت کاربران در توسعه آن است. حتی اگر برنامهنویس حرفهای نیستید، میتوانید به روشهای مختلفی به این پروژه کمک کنید. گزارش باگها، پیشنهاد ویژگیهای جدید، بهبود مستندات و نوشتن آزمونهای خودکار از جمله راههای مشارکت هستند. برای توسعهدهندگان پایتون، فرآیند مشارکت شامل fork کردن مخزن GitHub، ایجاد تغییرات و ارسال pull request است. پیش از شروع مشارکت، مطالعه راهنمای مشارکتکنندگان و آشنایی با استانداردهای کدنویسی پروژه توصیه میشود. مشارکت در پروژههای متنبازی مانند بایوپایتون، علاوه بر کمک به جامعه علمی، فرصتی برای یادگیری و گسترش مهارتهای برنامهنویسی است.
Biopython یک کتابخانه جامع برای تحلیلهای زیستی در محیط پایتون است که طیف گستردهای از ابزارها و توابع را برای متخصصان بیوانفورماتیک فراهم میکند. از مهمترین قابلیتهای این کتابخانه میتوان به پردازش توالیها، خواندن و نوشتن فرمتهای مختلف فایلهای زیستی، دسترسی به دیتابیسهای آنلاین، آنالیز فیلوژنتیک، تحلیل ساختار پروتئین و پشتیبانی از ابزارهای خارجی اشاره کرد. این قابلیتها، Biopython را به ابزاری کارآمد برای طیف وسیعی از کاربردها از جمله مطالعات تکاملی، طراحی دارو، ژنومیک، پروتئومیک و آنالیز دادههای توالییابی تبدیل کرده است. با یادگیری Biopython، محققان میتوانند فرآیندهای تحلیلی خود را خودکارسازی کنند، بر چالشهای محاسباتی غلبه کرده و به نتایج علمی دقیقتر و سریعتر دست یابند. Biolinux در کنار Biopython میتواند نوید بخش دورنمایی روشن برای بیوانفورماتیک باشد.
اگر تجربه استفاده از بایوپایتون رو دارید خوشحال میشیم توی بخش نظرات تجربیات خودتون رو با ما و همکارانتون به اشتراک بگذارید.
Biopython (بایوپایتون) یک کتابخانه متنباز برای زبان برنامهنویسی پایتون است که ابزارهای متنوعی برای تحلیل دادههای زیستی ارائه میدهد. این کتابخانه، امکاناتی مانند کار با توالیهای DNA، RNA و پروتئین، ساخت درختهای فیلوژنتیک، اتصال به دیتابیسهای زیستی و پردازش فایلهای زیستی مانند FASTA و PDB را در اختیار محققان قرار میدهد.
Biopython ابزارهای قدرتمندی برای تحلیل دادههای زیستی ارائه میدهد، اما برای دادههای بسیار حجیم مانند دادههای NGS بهتر است از ترکیب بایوپایتون با کتابخانههایی مانند NumPy یا pandas استفاده کنید. همچنین میتوانید با استفاده از پردازش موازی و بهینهسازی کد، عملکرد آن را افزایش دهید.
خیر، Biopython علاوه بر تحلیل توالیها، قابلیتهای دیگری مانند ساخت درختهای فیلوژنتیک، کار با ساختارهای سهبعدی پروتئین، اتصال به دیتابیسهای زیستی مانند NCBI و PDB و پردازش فایلهای زیستی در فرمتهای مختلف را ارائه میدهد.
با استفاده از ماژول Bio.Entrez میتوانید به پایگاه دادههای NCBI متصل شوید و دادههای مورد نیاز خود را جستجو، دریافت و تحلیل کنید. همچنین، ماژول Bio.PDB امکان دسترسی به دادههای ساختاری پروتئینها در پایگاه PDB را فراهم میکند.

تیم تولید محتوای گروه بیوانفورماتیک وانیار در تلاش است تا بهترین آموزشهای کوتاه در زمینه بیوانفورماتیک و زیستشناسی را تهیه نماید. صحت محتوای این صفحه توسط کارشناسان گروه بیوانفورماتیک وانیار بررسی شده است.



عضویت در مجله وانیار
جدید ترین مقالات در ایمیل شما!
با عضویت در مجله بیوانفورماتیک وانیار ، برترین مقالات را در ایمیل خود دریافت کنید.
سلام، وقت بخیر.
چطور میتونیم بهتون کمک کنیم؟ 🤓
تیم ما آماده پاسخگویی به سوالات شماست.
پشتیبانی 24 ساعته در 7 روز هفته.






