دوره NGS منتشر شد …

BLAST (ابزار جستجوی همردیفی محلی پایه) یکی از مهمترین و پرکاربردترین ابزارهای بیوانفورماتیکی است که برای جستجو و مقایسه توالیهای زیستی در پایگاههای داده استفاده میشود. این ابزار با سرعت و دقت بالایی که دارد، توانسته است جایگاه ویژهای در پژوهشهای زیستی و پزشکی به دست آورد. BLAST به محققان این امکان را میدهد که به سرعت توالیهای DNA ،RNA و پروتئینها را با توالیهای مرجع مقایسه کنند و شباهتها یا تفاوتهای آنها را شناسایی نمایند. کاربردهای BLAST از شناسایی ژنها و پروتئینها تا بررسی تکامل و ارتباطات فیلوژنتیکی موجودات زنده را شامل میشود.
BLAST در دهه 1990 توسط آلچول و همکارانش در موسسه ملی سلامت آمریکا (NIH) توسعه داده شد. هدف اصلی آن، ارائه یک الگوریتم سریع برای جستجو و همردیفی توالیها بود، زیرا روشهای سنتی مانند همردیفیهای سراسری(Global alignment)، به زمان محاسباتی بالایی نیاز داشتند و برای مجموعه دادههای بزرگ مقرونبهصرفه نبودند.
BLAST با معرفی الگوریتمهای جستجوی محلی که بر اساس شناسایی نواحی مشابه در توالیها است، موفق شد تا فرآیند جستجو را بهشدت سریعتر کند. این ابزار در طول سالها بهروز شده و نسخههای مختلفی از آن، شامل BLASTn، BLASTp، و BLASTx برای کاربردهای متنوع ایجاد شدهاند.
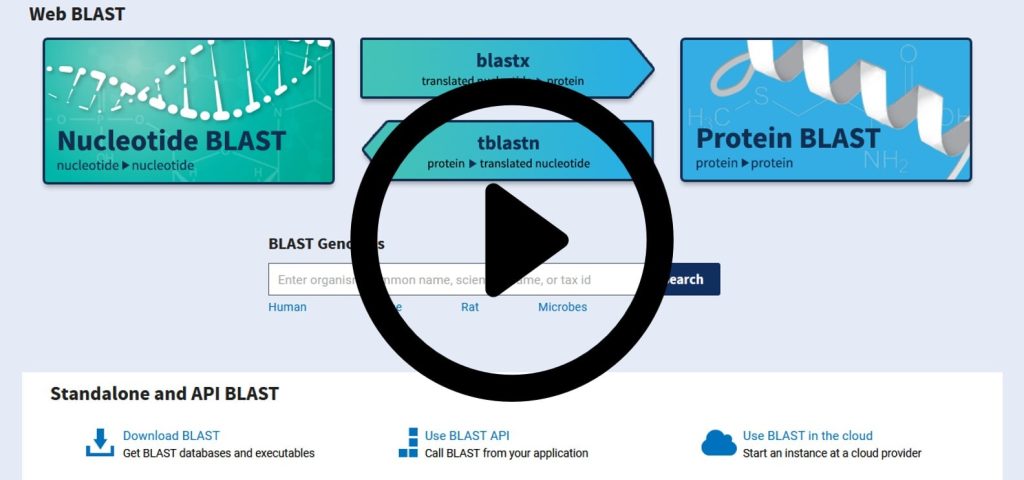
BLAST شامل چندین نسخه متفاوت است که هر کدام برای مقایسه نوع خاصی از توالیها به کار میروند. انواع مختلف بلاست از سایت NCBI در دسترس است. برخی از مهمترین انواع آن عبارتاند از:
این نسخه از BLAST برای مقایسه توالیهای نوکلئوتیدی (RNA و DNA) طراحی شده است. BLASTn به پژوهشگران این امکان را میدهد تا توالیهای DNA یا RNA را با سایر توالیهای نوکلئوتیدی مقایسه کرده و همردیفیهای احتمالی را شناسایی کنند.
این نوع از BLAST برای مقایسه توالیهای پروتئینی استفاده میشود. BLASTp با تمرکز بر توالیهای آمینواسیدی، پروتئینهای همساختاری را شناسایی کرده و شباهتهای آنها را با سایر پروتئینهای موجود در پایگاه داده ارزیابی میکند.
این نسخه، توالیهای نوکلئوتیدی را با ترجمههای احتمالی پروتئینی مقایسه میکند. BLASTx به محققان کمک میکند تا توالیهای کدگذاری نشده را به توالیهای پروتئینی ترجمه کنند و سپس با سایر توالیهای پروتئینی مقایسه نمایند.
در این نوع BLAST، توالیهای پروتئینی با توالیهای نوکلئوتیدی ترجمهشده مقایسه میشوند. tBLASTn برای جستجوی پروتئینها در پایگاههای داده ژنومی به کار میرود.
این نسخه، توالیهای نوکلئوتیدی ترجمهشده را با هم مقایسه میکند و برای جستجوهای عمیقتر در توالیهای ژنومی استفاده میشود.
هر یک از این نسخهها برای اهداف خاصی طراحی شده و با ارائه نتایج دقیق، کاربرد گستردهای در پژوهشهای ژنتیکی، پزشکی و بیوتکنولوژی دارند.
BLAST از الگوریتم جستجوی محلی استفاده میکند که به جای همردیفی سراسری، به دنبال نواحی مشابه در توالیها میگردد. این روش از چند مرحله اصلی تشکیل شده است:
در این مرحله، BLAST کلمات یا قطعات کوتاهی از توالی ورودی را شناسایی میکند. این کلمات کوتاه، به طور معمول سه یا چهار حرفی در توالیهای پروتئینی یا نوکلئوتیدی هستند که با جستجو در پایگاه داده، به سرعت مقایسه میشوند.
پس از یافتن نواحی مشابه با کلمات اولیه در این مرحله، BLAST کلمات اولیه را با استفاده از معیارهای امتیازدهی گسترش میدهد و به دنبال نواحی همردیفی بیشتر میگردد. این مرحله به شناسایی شباهتهای بزرگتر و یافتن توالیهای مشابه کمک میکند.
در نهایت، BLAST با استفاده از یک سیستم امتیازدهی به همردیفیهای بهدستآمده ارزشدهی میکند. این امتیازدهی بر اساس مدلهای جایگزینی آمینواسیدها و نوکلئوتید مانند ماتریس PAM و BLOSUM انجام میشود و به پژوهشگران این امکان را میدهد تا توالیهای همساختاری یا ژنهای مشابه را شناسایی کنند.
این روش کارآمد و بهینه، BLAST را به ابزاری محبوب و سریع برای تجزیهوتحلیل توالیهای ژنتیکی تبدیل کرده است.
باید در نظر داشت که کاربرد BLAST در زیستشناسی همچون کاربرد میکروسکوپ گسترده است و هر محقق متناسب با نیاز پروژه خود میتواند از این ابزار قدرتمند استفاده نماید. با توجه به این موضوع که تقریبا اصول استفاده از BLAST در پروژههای مختلف بیوانفورماتیک شبیه به هم میباشد، در ادامه با استفاده از یک مثال عملی شیوه استفاده از ابزار BLAST را توضیح خواهیم داد.
ابتدا از طریق لینک فوق به ابزار BLAST وارد میشویم:
سپس متناسب با نوع توالی و هدف پروژه ابزار مناسب بلاست را انتخاب خواهیم نمود در این جا ما قصد داریم یک توالی نوکلئوتیدی حاصل از یک ویروس نامعلوم را به منظور تعیین گونه ویروسی BLAST کنیم. بنابراین ابزار BLASTn را انتخاب خواهیم نمود.
توالی مورد نظر را به فرمت FASTA در کادر زیر وارد میکنیم. همچنین میتوانیم فایل توالی مورد نظر را با زدن گزینه Browse از روی سیستم به صورت لوکال انتخاب کنیم. یک نام برای پروژه بلاست خود انتخاب میکنیم.
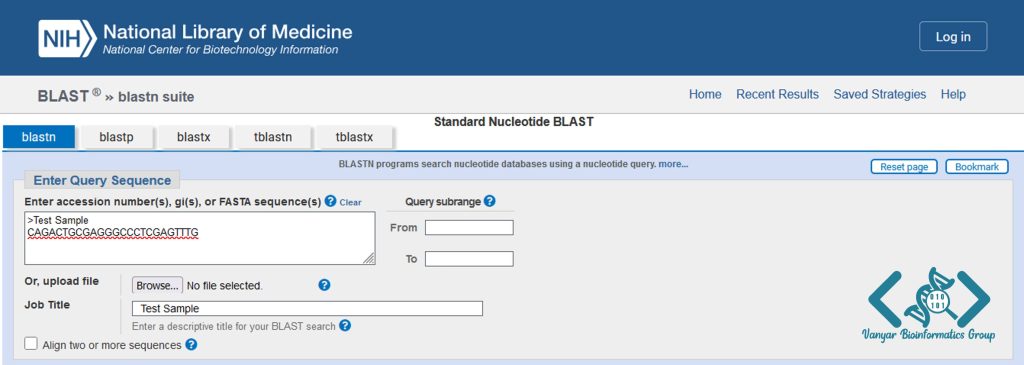
در کادر Choose Search Set پایگاه داده خود را بر روی Core nucleotide database (Core_nt) تنظیم میکنیم تا نتایج تکراری را برای ما حذف نماید.
در بخش Program Selection نوع برنامه را بر روی Highly similar sequences (megablast) قرار میدهیم این برنامه سرعت و دقت مناسبی را برای جستجوی حجم بالایی از دادهها دارد.
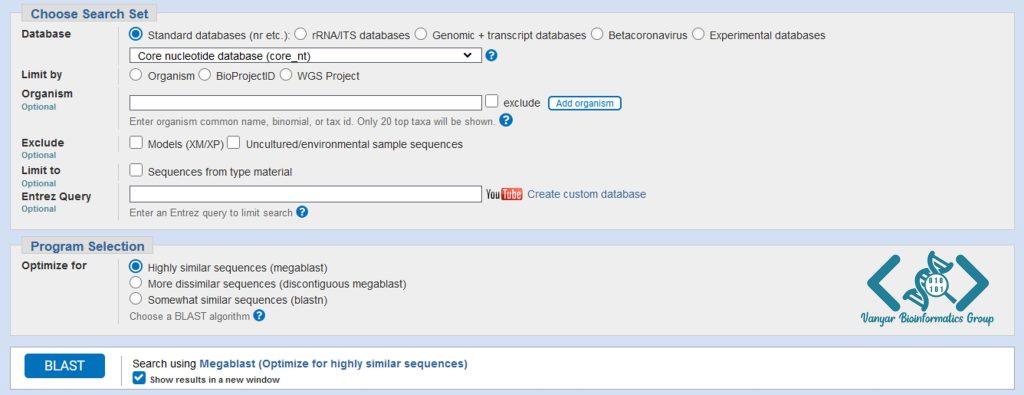
به منظور حفظ صفحه تنظیمات جستجو و برای تغییر در گزینههای تنظیمی بلاست پس از جستجوی توالی، گزینهی Show results in a new window را انتخاب میکنیم تا نتایج در یک زبانهی جدید نمایش داده شود، با این کار در صورت نیاز میتوانید تنظیمات جستجوی بلاست را پس از ظاهر شدن نتایج تغییر دهید و جستجو را دوباره اجرا نمایید.
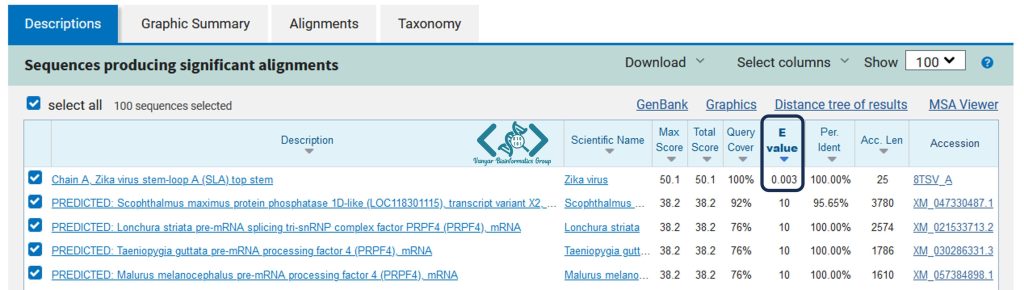
متناسب با طول توالی و حجم پایگاه داده مورد جستجو، پس از گذشت زمان مشخص نتایج جستجوی BLAST به شما نمایش داده خواهد شد.
مقدار E یا E-Value (Expected Value) یکی از مهمترین پارامترها در تحلیل نتایج BLAST است و به پژوهشگران کمک میکند تا میزان معنادار بودن شباهتهای مشاهده شده بین توالیها را ارزیابی کنند. E-Value بیانگر تعداد همردیفیهای مشابهی است که بهطور تصادفی در پایگاه دادهی جستجو شده قابل انتظار است و نشان میدهد که احتمال دارد نتیجه بهدستآمده، به دلیل شباهتهای تصادفی و نه واقعی، معنادار به نظر برسد.
به بیان سادهتر، هرچه مقدار E-Value کمتر باشد، احتمال تصادفی بودن همردیفی کمتر است و شباهت مشاهده شده معنادارتر تلقی میشود. به عنوان مثال، یک E-Value بسیار کوچک (مثلاً 1e-10 یا 0.0000000001) نشان میدهد که احتمال این که شباهت مشاهده شده به طور تصادفی رخ داده باشد بسیار ناچیز است و بنابراین، همردیفی معنادارتر و معتبرتر است.
E Value به عوامل متعددی بستگی دارد، از جمله:
به طور کلی، رابطه بین نمره همردیفی و E-Value به این صورت است که هرچه نمره همردیفی بیشتر باشد، E-Value کمتر خواهد بود، و برعکس.
در تحلیل نتایج BLAST، معمولاً از یک مقدار آستانه برای E Value استفاده میشود. به طور معمول، پژوهشگران مقدار 0.01 یا کمتر را به عنوان معیار معناداری در نظر میگیرند. بنابراین، اگر مقدار E-Value کمتر از این حد باشد، شباهت به عنوان یک همردیفی معنادار و واقعی در نظر گرفته میشود.
در مجموع، E Value به پژوهشگران کمک میکند تا نتایج BLAST را غربال کنند و همردیفیهای معنادار را از همردیفیهای احتمالی تصادفی تفکیک نمایند، که این امر در تجزیهوتحلیل دقیق و علمی توالیها بسیار حیاتی است.
BLAST بهعنوان یک ابزار بیوانفورماتیکی توانسته است نقش بسیار مهمی در پژوهشهای زیستی و پزشکی ایفا کند. برخی از کاربردهای اصلی آن عبارتاند از:
BLAST به پژوهشگران کمک میکند تا ژنها و پروتئینهای همولوگ را شناسایی کرده و تفاوتهای ژنتیکی بین گونهها را بررسی کنند.
در تحقیقات پزشکی، BLAST به شناسایی ژنهای دخیل در بیماریها و نیز شناسایی پاتوژنهای مختلف کمک میکند. با مقایسه توالیهای پاتوژنها با توالیهای شناختهشده، امکان شناسایی عاملین بیماریزا فراهم میشود.
BLAST امکان بررسی ارتباطات فیلوژنتیکی و تحلیل تکاملی را فراهم میکند. پژوهشگران میتوانند با استفاده از این ابزار، توالیهای ژنتیکی مختلف را با هم مقایسه کرده و شباهتها و تفاوتهای تکاملی را شناسایی کنند.
BLAST با شناسایی توالیهای پروتئینی مشابه، به پیشبینی ساختار سهبعدی پروتئینها کمک میکند. این امر در طراحی داروها و تحقیقات زیستفناوری اهمیت ویژهای دارد. با انجام بلاست در پایگاه UniProt میتوان عملکرد پروتئینها تازه توالییابی شده را تعیین کرد.
دوره بیوانفورماتیک پروتئین: یک فرصت طلایی برای فراگیری مهارتهای مختلف در حوزه بیوانفورماتیک پروتئینها است.

BLAST به پژوهشگران اجازه میدهد تا توالیهای نوین ژنومی و پروتئینی را شناسایی کرده و آنها را با توالیهای موجود مقایسه کنند. این قابلیت در پروژههای ژنومیک و مطالعه تنوع زیستی کاربرد فراوانی دارد.
یکی از کاربردهای تخصصی BLAST، طراحی پرایمر اختصاصی است. همچنین میتوان با بلاست کیفیت و اختصاصیت پرایمرهای طراحی شده را بررسی کرد که یک نوع PCR به صورت in silico است.
دوره جامع طراحی پرایمر: آموزش طراحی پرایمر برای تکنیکهای مختلف PCR با ابزارهای پرکاربرد در طراحی پرایمر.

BLAST یکی از مهمترین ابزارهای بیوانفورماتیکی است که به پژوهشگران علوم زیستی و پزشکی امکان تجزیهوتحلیل سریع و دقیق توالیهای ژنتیکی را میدهد. از شناسایی ژنها و پروتئینها گرفته تا مطالعات تکاملی و طراحی دارو، BLAST کاربردهای گستردهای دارد. این ابزار به مرور زمان با توسعه نسخههای مختلف، توانسته است نیازهای پژوهشی در زمینههای متنوع را برطرف کند و به یکی از ابزارهای اساسی در تحقیقات ژنومی و بیوتکنولوژی تبدیل شود.
BLAST به ابزاری قوی و پر سرعت برای جستجوی توالی در پایگاههای مختلف تبدیل شده است، این امر سبب شده که کاربردهای بلاست گسترده باشند. پیشرفتهای اخیر در الگوریتمها و بهبودهای نرمافزاری BLAST، این ابزار را بهعنوان یکی از ابزارهای ضروری برای محققان حفظ کرده و تواناییهای آن در آینده نیز به احتمال زیاد افزایش خواهد یافت. اگر تجربه استفاده از ابزار BLAST رو داری خوشحال میشیم توی کامنتها از تجربیات خودت برامون بگی.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., & Lipman, D. J. (1990). Basic local alignment search tool. Journal of Molecular Biology, 215(3), 403-410.
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., & Madden, T. L. (2009). BLAST+: architecture and applications. BMC Bioinformatics, 10(1), 421.
BLAST (Basic Local Alignment Search Tool) یک ابزار بیوانفورماتیکی است که برای مقایسه توالیهای DNA ،RNA و پروتئینها با توالیهای موجود در پایگاههای داده استفاده میشود.
BLAST برای شناسایی ژنها، پروتئینهای مشابه، مطالعات تکاملی، طراحی پرایمر و بررسی شباهتهای ژنتیکی استفاده میشود.
بله BLAST یک ابزار کاملا رایگان است و در دسترس تمامی محققان قرار دارد.
E-Value (Expected Value) نشاندهنده تعداد همردیفیهایی است که به طور تصادفی در پایگاه داده قابل انتظار است. هرچه مقدار E-Value کمتر باشد، شباهت مشاهده شده معنادارتر است. معمولاً مقدار 0.01 یا کمتر به عنوان یک همردیفی معنادار در نظر گرفته میشود.

تیم تولید محتوای گروه بیوانفورماتیک وانیار در تلاش است تا بهترین آموزشهای کوتاه در زمینه بیوانفورماتیک و زیستشناسی را تهیه نماید. صحت محتوای این صفحه توسط کارشناسان گروه بیوانفورماتیک وانیار بررسی شده است.



عضویت در مجله وانیار
جدید ترین مقالات در ایمیل شما!
با عضویت در مجله بیوانفورماتیک وانیار ، برترین مقالات را در ایمیل خود دریافت کنید.
سلام، وقت بخیر.
چطور میتونیم بهتون کمک کنیم؟ 🤓
تیم ما آماده پاسخگویی به سوالات شماست.
پشتیبانی 24 ساعته در 7 روز هفته.