دوره NGS منتشر شد …

FASTA یکی از مفاهیم بنیادین در علم بیوانفورماتیک است که دارای دو جنبه متمایز میباشد. از یک سو، FASTA به عنوان یک فرمت استاندارد برای ذخیرهسازی دادههای توالی بیولوژیکی (نظیر DNA ،RNA و پروتئین) شناخته میشود، و از سوی دیگر، به عنوان یک الگوریتم قدرتمند برای جستجوی شباهت بین توالیهای زیستی کاربرد دارد. این دوگانگی گاهی موجب سردرگمی محققان تازهکار در این حوزه میشود، اما درک صحیح هر دو جنبه برای کار موثر در حوزههای مختلف زیستشناسی محاسباتی ضروری است. در این مقاله، به بررسی جامع هر دو جنبه FASTA میپردازیم تا دید کاملی از کاربردها و اهمیت آن در دنیای امروز علوم زیستی به دست آوریم.
فَست-A در اوایل دهه 1980 توسط دیوید لیپمن و ویلیام پیرسون در دانشگاه ویرجینیا توسعه یافت. نام این سیستم مخفف “Fast-All” است که اشاره به توانایی آن در جستجوی سریع در تمام توالیهای موجود در دیتابیسها دارد. در ابتدا، FASTA به عنوان یک الگوریتم جستجوی توالی معرفی شد و پیشرفت قابل توجهی نسبت به روشهای جستجوی توالی قبلی محسوب میشد. با گذشت زمان، فرمت فایل سادهای که برای ورودی و خروجی این الگوریتم استفاده میشد، به یک استاندارد جهانی تبدیل شد.
از زمان معرفی، FASTA چندین بار بهبود یافته و نسخههای مختلفی از آن عرضه شده است. نسخه اولیه FASTP نام داشت که مخصوص مقایسه توالیهای پروتئینی بود، اما نسخههای بعدی قابلیتهای گستردهتری از جمله مقایسه توالیهای DNA و مقایسههای ترجمهای را ارائه دادند. امروزه، علیرغم ظهور الگوریتمهای جدیدتر مانند BLAST، الگوریتم FASTA همچنان بهدلیل دقت بالا و سادگی استفاده، جایگاه ویژهای در جامعه زیستشناسی محاسباتی دارد.
فرمت فایل فستا یکی از سادهترین و پرکاربردترین فرمتها برای ذخیره دادههای توالی بیولوژیکی است. این فرمت از دو بخش اصلی تشکیل شده: خط توصیف (header) که با علامت بزرگتر (<) شروع میشود و حاوی اطلاعات مربوط به توالی است، و بدنه اصلی که شامل خود توالی (نوکلئوتیدی یا آمینواسیدی) است. توالی میتواند در یک یا چند خط ارائه شود، اما خط توصیف همیشه در یک خط واحد قرار میگیرد.
مثالی از یک فایل فستا حاوی یک توالی DNA به صورت زیر است:
>gi|186681228|ref|YP_001864424.1| phycoerythrobilin synthase [Nostoc sp. PCC 7120]
ATGAGTGTCAACGTTGGTCAAGCCATCGGCACAGGCTTGGGATCAGCCGATCGTCACGGCGGTCGTCAGGATATC
GCCCTGGATCACCTGAAGGCGATCGCCCAGAGCAAGAGCGGCAAGGAAACGATCCTCACAGGCAAGGCGATCTGC
CGCCGCATCACCGAGTTGAAGAAGGCCGGTGTCAACGGCGTCAAAGACATGATCGCCCGTCACGGTGTTGACCAC
ACCAACACCTGGAACACCAAGGGCAAGACCGAGCTGGTGGTCAACCTCGGCGACGATGCCACCGGCGGCATCTGC
ACGATGGTGGAGAAGTTCTACAAGCTGATCGGCATTGACGACGTCAGCGاز مزایای اصلی فرمت فستا عبارتند از: سادگی ساختار، خوانایی بالا برای انسان، حجم کم فایل، سازگاری با اغلب نرمافزارهای بیوانفورماتیک، و انعطافپذیری در ذخیره انواع مختلف توالیها. این ویژگیها باعث شده فستا به یک استاندارد جهانی برای تبادل دادههای توالی بیولوژیکی تبدیل شود، به نحوی که تقریباً تمام دیتابیسهای توالی امکان دانلود دیتا با این فرمت را فراهم میکنند.
پیشنهاد: در دوره بیوانفورماتیک عمومی و کاربردی، الگوریتم FASTA به صورت کاربردی آموزش داده شده است.

الگوریتم فست-اِی یک روش کارآمد برای جستجوی شباهت بین توالیهای بیولوژیکی است که بر اساس یافتن نواحی کوتاه مشابه بین توالیهای مورد مقایسه عمل میکند. این الگوریتم در چهار مرحله اصلی کار میکند:
(1) شناسایی مناطق با تشابه بالا با استفاده از روش k-tuple
(2) شناسایی 10 ناحیه با بالاترین نمره مشابهت
(3) بررسی دقیقتر نواحی یافتهشده با استفاده از ماتریسهای امتیازدهی (مانند PAM یا BLOSUM)
(4) ترکیب نتایج برای ساخت یک همترازی نهایی.
مزیت اصلی الگوریتم فَست اِی نسبت به روشهای اولیه، استفاده از روش k-tuple است که سرعت جستجو را به طور قابل توجهی افزایش میدهد. در این روش، ابتدا توالیها به زیرتوالیهای کوتاه به طول k (معمولاً 1 یا 2 برای پروتئینها و 4 یا 6 برای DNA) تقسیم میشوند و مکانهای مشترک این زیرتوالیها شناسایی میشوند. این رویکرد باعث میشود بخش زیادی از مقایسههای غیرضروری حذف شود و در نتیجه سرعت الگوریتم افزایش یابد.
در مقایسه با BLAST، الگوریتم فست-اِی معمولاً از دقت بیشتری برخوردار است اما سرعت کمتری دارد. نسخههای مختلف این الگوریتم برای انواع مختلف جستجوها توسعه یافتهاند، از جمله FASTP برای مقایسه پروتئینها، FASTX و FASTY برای مقایسههای ترجمهای، و FASTA3 که نسخه بهبودیافته و کاملتری از این الگوریتم است. امروزه، علیرغم محبوبیت بیشتر BLAST، الگوریتم FASTA همچنان به عنوان یک ابزار ارزشمند برای تحلیلهای دقیقتر توالی مورد استفاده قرار میگیرد.
برای استفاده از FASTA، ابتدا باید فایلهای توالی خود را در فرمت فستا (یا سایر فرمتهای قابلقبول، مانند GenBank و GFF3) آماده کنید. هر توالی باید با یک خط توصیف که با علامت < شروع میشود آغاز شود، و در خطوط بعدی خود توالی قرار گیرد. برای نتایج بهتر، توصیه میشود از شناسههای معنادار و توصیفهای مختصر در خط عنوان استفاده کنید. توجه داشته باشید که طول هر خط توالی میتواند متغیر باشد، اما معمولاً برای خوانایی بهتر، هر خط حداکثر 80 کاراکتر در نظر گرفته میشود.
پس از آمادهسازی فایلهای توالی، میتوانید جستجو را با استفاده از GUI وبسایت FASTA اجرا کنید.
برای مثال، برای : پیدا کردن همولوگهای هموگلوبین-بتای انسانی (UniProt P68871)، مراحل زیر را دنبال میکنیم:
مرورگر خود را باز کرده و به صفحهی FASTA در EMBL-EBI وارد شوید.
در بخش ”Databases” از منوی کشویی، روی UniProtKB/Swiss-Prot یا هر دیتابیس پروتئینی دیگری که مدنظرتان است، کلیک کنید.
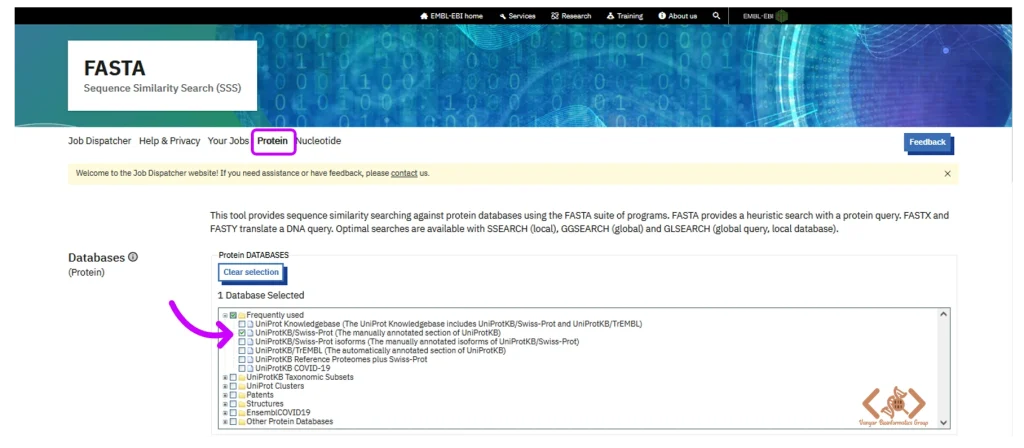
نوع توالی (DNA | RNA | Protein) را انتخاب کرده و در کادر زیر آن، توالی query (توالی مورد بررسی خود، در اینجا هموگلوبین β انسانی) را وارد کنید. از قسمت Browse هم میتوانید فایل توالی را ضمیمه کنید.
از بخش Program نیز برنامه جستجو را تعیین کنید، که ما قصد داریم توالی پروتئینی را در دیتابیس پروتئینی جستجو کنیم و FASTA را انتخاب میکنیم.
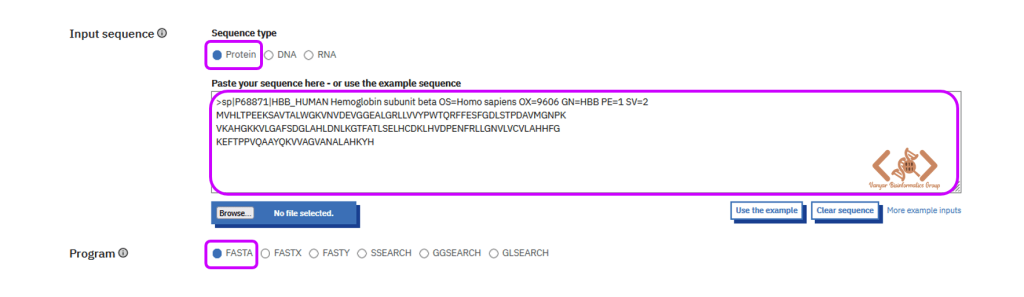
در این مرحله میتوانید پارامترهایی همچون موارد زیر را تنظیم کنید:
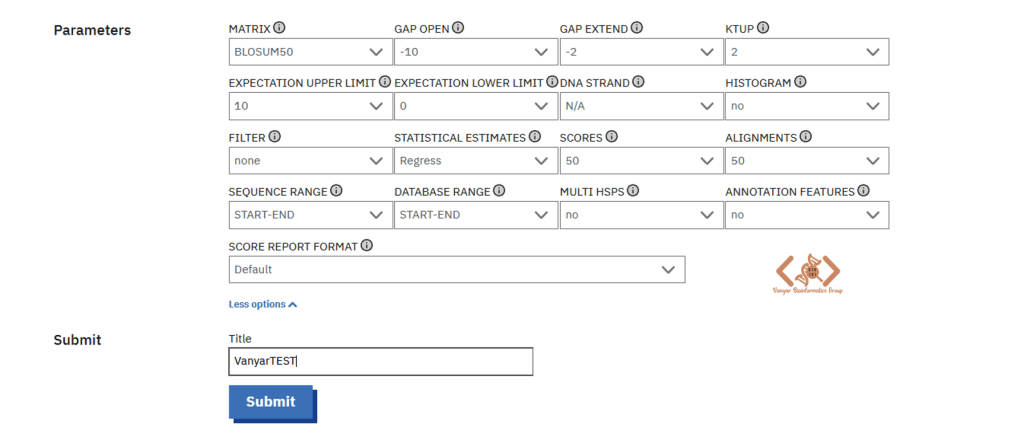
MATRIX : ماتریس جایگزینی برای امتیازدهی جایگشتهای جفت نوکلئوتیدی/آمینواسیدی (مثلاً BLOSUM50, PAM250)
GAP OPEN : جریمهای که برای بازشدن هر گپ به امتیاز همترازی اضافه میشود (مقدار منفی)
GAP EXTEND : جریمهای که برای هر نوکلئوتید/آمینواسید اضافی درون یک گپ اعمال میشود.
KTUP : طول کلمه (word size) برای جستجوی k-tupleاولیه؛ عدد کوچکتر حساسیت را افزایش میدهد.
STATISTICAL ESTIMATES : روش برآورد آماری E-value، مثلاً regress vs. shuffle
بعد از نامگذاری این ران به اسم دلخواه، روی دکمهی Submit کلیک کنید تا جستجو آغاز شود. پس از اتمام اجرا، با کلیک بر View Results، به صفحهی نتایج هدایت میشوید.
دربخش Summary Table فهرستی از توالیهای همولوگ را به ترتیب E-value، نرخ همسانی (identity %) و امتیاز (score) مشاهده میکنید (تصویر پایین سمت چپ).
با ورود به تب Tool Output هم میتوانید الاینمنت توالیها را با جزئیات BLOSUM50، نواحی gap و پوشش توالی ببینید (تصویر پایین سمت راست).

Hit های بالاتر، معمولاً نسخههای β-هموگلوبین در دیگر پستانداران هستند؛
E-value پایین نشاندهندهی شباهت بسیار بالا و احتمال همولوژی واقعی است؛
%Identity و %Similarity به شما میگویند چند درصد از باقیماندهها دقیقاً یکسان یا شبیه هستند.
با این روش میتوانید برای هر توالی دلخواه، همولوگهای نزدیک و دور را در دیتابیسهای متنوع شناسایی کنید.
در تفسیر نتایج، باید به چند پارامتر کلیدی توجه کرد: نمره همترازی (bit score)، E-value (که احتمال تصادفی بودن تشابه را نشان میدهد)، درصد یکسانی (identity) و پوشش توالی (coverage). E-value کمتر (نزدیک به صفر) نشاندهنده تشابه معنادارتر است. معمولاً E-value کمتر از 0.001 برای شناسایی ارتباطات معنادار بین توالیها استفاده میشود. نتایج معمولاً به صورت لیستی از همترازیها به ترتیب نزولی نمره نمایش داده میشوند.
🟥 یکی از اشتباهات رایج، تفسیر نادرست E-value یا تکیه بیش از حد بر درصد یکسانی بدون در نظر گرفتن طول همترازی است. برای رفع این مشکلات، بهتر است همیشه ترکیبی از پارامترها را در نظر بگیرید و نتایج را با دقت بررسی کنید. همچنین، در صورت امکان، نتایج را با سایر روشهای تحلیلی تأیید کنید.
الگوریتم و بستهی نرمافزاری FASTA یکی از نخستین و مؤثرترین ابزارهای جستجوی شباهت توالی در بیوانفورماتیک محسوب میشود که همچنان نقش مهمی در تحلیل دادههای زیستی ایفا میکند. این ابزار با تکیه بر تطبیق جفتبهجفت (pairwise alignment) توالیها، در بسیاری از حوزههای پژوهشی علوم زیستی کاربرد دارد. در ادامه به چند مورد از رایجترین کاربردهای FASTA اشاره میکنیم:
یکی از کاربردهای کلیدی FASTA، شناسایی توالیهای همولوگ بین گونههای مختلف است. محققان میتوانند با استفاده از این ابزار، ژنهای ارتولوگ (orthologs) و پارالوگ (paralogs) را تشخیص دهند و از آن برای بازسازی روابط تکاملی و درختهای فیلوژنتیک بهره ببرند. این تحلیلها در درک تکامل ژنها، بررسی انتخاب طبیعی و شناسایی رویدادهای Gene Duplication اهمیت دارند. بهعلاوه، خروجیهای FASTA معمولاً بهعنوان ورودی نرمافزارهایی مانند Clustal، OrthoFinder و ابزارهای ترسیم درخت استفاده میشوند.
FASTA در ترکیب با پایگاههایی مانند Pfam و InterPro، در شناسایی دومینهای عملکردی، سایتهای فعال و موتیفهای حفاظتشده پروتئینها نقش دارد. این آنالیزها به درک عملکرد مولکولی پروتئینها کمک میکند و بهویژه در پروژههای پروتئومیک، طراحی دارو و مهندسی پروتئین اهمیت دارند. نسخههای جدیدتر FASTA امکان مشاهده مستقیم دومینها و ویژگیهای ساختاری را نیز فراهم کردهاند.
در پروژههای مدلسازی ساختار پروتئین، یافتن قالب ساختاری (template) مشابه، اولین و حیاتیترین گام است. فست اِی با مقایسه توالی پروتئین هدف با توالیهای شناختهشده موجود در پایگاههایی مانند PDB، پروتئینهای مشابه را پیشنهاد میدهد. سرورهای مدلسازی مانند SWISS-MODEL و Modeller از خروجی FASTA برای ساخت مدلهای همولوژی بهره میبرند.
الگوریتم FASTA میتواند برای تشخیص گونه از طریق DNA Barcoding بهکار رود؛ با قرار دادن توالی بارکد (مثلاً COI در جانوران) و مقایسهٔ جفتبهجفت آن با کتابخانههای مرجع، میتوان گونهٔ نمونه را تعیین کرد.
FASTA و BLAST دو الگوریتم اصلی و پرکاربرد در جستجوی شباهت توالی هستند که هر کدام نقاط قوت و ضعف خاص خود را دارند. BLAST (Basic Local Alignment Search Tool) که در سال 1990 معرفی شد، سرعت بالاتری نسبت به FASTA دارد و به همین دلیل محبوبیت بیشتری در میان کاربران پیدا کرده است. BLAST از روش “seed-and-extend” استفاده میکند که در آن ابتدا بذرهای کوچک (words) یافت میشوند و سپس در جهات مختلف گسترش مییابند. در مقابل، FASTA با استفاده از روش k-tuple، ابتدا مناطق با تشابه بالا را شناسایی و سپس آنها را با دقت بیشتری بررسی میکند.
از نظر دقت، FASTA به ویژه در شناسایی همولوگهای دور (توالیهایی با تشابه کمتر) عملکرد بهتری دارد، زیرا از روشهای حساستری برای امتیازدهی به همترازیها استفاده میکند. در مقابل، BLAST برای جستجوهای سریع در پایگاهدادههای بزرگ مناسبتر است. تفاوت دیگر این است که فست-اِی امکان تنظیم دقیقتر پارامترهای جستجو را فراهم میکند که برای کاربردهای تخصصی مفید است، در حالی که BLAST رابط کاربری سادهتری دارد که برای کاربران تازهکار مناسبتر است.
در کنار این دو الگوریتم کلاسیک، ابزارهای مدرنتری نیز توسعه یافتهاند که سرعت و دقت بیشتری را ارائه میدهند. برای مثال، DIAMOND برای جستجوهای پروتئینی تا 20000 برابر سریعتر از BLAST عمل میکند، و MMseqs2 برای جستجوهای حساس و در مقیاس بزرگ بهینهسازی شده است. همچنین، الگوریتمهای مبتنی بر یادگیری عمیق مانند AlphaFold-MSA و ESM-MSA دقت بیشتری در شناسایی روابط تکاملی دور دارند.
با وجود این پیشرفتها، FASTA همچنان در برخی حوزهها مزایای خاص خود را دارد. به طور خاص، هنگامی که نیاز به کنترل دقیق بر پارامترهای جستجو، حساسیت بالا در شناسایی همولوگهای دور، یا تفسیر دقیق نتایج آماری وجود دارد، FASTA میتواند انتخاب مناسبی باشد. علاوه بر این، سادگی و قابلیت انتقال کد FASTA آن را برای ادغام در پایپلاینهای تحلیلی سفارشی مناسب میسازد.
در سالهای اخیر، نسخههای جدید الگوریتم فَست اِی با هدف افزایش سرعت، دقت و کارایی توسعه یافتهاند. یکی از مهمترین پیشرفتها، پیادهسازی محاسبات موازی در FASTA36 است که از چندین هسته پردازشی برای اجرای همزمان جستجوها استفاده میکند. این ویژگی به خصوص در جستجوهای گسترده در پایگاهدادههای بزرگ، سرعت را به طور قابل توجهی افزایش میدهد. علاوه بر این، بهینهسازیهای الگوریتمی مانند استفاده از ساختارهای داده کارآمدتر و روشهای جستجوی بهینهتر، باعث کاهش مصرف حافظه و افزایش سرعت پردازش شده است.
پیشرفت دیگر، توسعه نسخههای خاص FASTA برای انواع مختلف جستجوهاست. برای مثال، FASTX و FASTY برای جستجوهای ترجمهای (مقایسه توالی DNA با پایگاه داده پروتئینی) بهینه هستند و FASTS و FASTF برای جستجوی توالیهای کوتاه پپتیدی طراحی شدهاند. همچنین، نسخههای تخصصی برای جستجوی موتیفهای خاص و تحلیلهای ساختاری توسعه یافتهاند که کاربردهای FASTA را گسترش دادهاند.
پیادهسازیهای مبتنی بر فناوریهای ابری نیز یکی دیگر از پیشرفتهای اخیر در زمینه FASTA است. این پیادهسازیها امکان اجرای جستجو روی منابع محاسباتی توزیعشده را فراهم میکنند که برای پردازش حجم عظیم دادههای توالی در عصر “omics” ضروری است. پلتفرمهایی مانند Galaxy و CyVerse رابطهای کاربرپسندی برای اجرای FASTA در محیط ابری ارائه میدهند که دسترسی به این ابزار را برای محققان بدون دانش فنی گسترده تسهیل میکند.
با این حال، FASTA همچنان با چالشهایی مواجه است. افزایش نمایی حجم دادههای توالی، نیاز به الگوریتمهای کارآمدتر را افزایش میدهد. همچنین، شناسایی روابط بسیار دور تکاملی که تشابه توالی اندکی دارند، همچنان یک چالش باقی مانده است. روشهای جدید مبتنی بر یادگیری عمیق و تحلیلهای چندبعدی توالی-ساختار در حال توسعه هستند که میتوانند این محدودیتها را برطرف کنند. با این حال، FASTA به عنوان یک پایه قوی و قابل اطمینان برای تحلیل توالیهای بیولوژیکی، همچنان نقش مهمی در پیشبرد تحقیقات زیستشناسی محاسباتی ایفا میکند.
پیشنهاد: Chromas چیست؟ آموزش و دانلود رایگان نرمافزار Chromas
برای استفاده بهینه از FASTA، درک صحیح هر دو جنبه آن (فرمت فایل و الگوریتم جستجو)، آشنایی با پارامترهای مختلف و نحوه تفسیر نتایج، و همچنین آگاهی از محدودیتها و مکملهای آن ضروری است. با توجه به رشد نمایی دادههای توالی و پیچیدگی روزافزون سؤالات زیستشناسی، ترکیب FASTA با سایر ابزارها و روشهای تحلیلی میتواند به شناخت عمیقتر سیستمهای بیولوژیکی کمک کند.
در نهایت، FASTA به عنوان یکی از قدیمیترین اما همچنان پرکاربردترین ابزارهای بیوانفورماتیک، نمونهای از اصول پایدار و اساسی در علم است که علیرغم پیشرفتهای سریع فناوری، ارزش و کاربرد خود را حفظ کرده است. آشنایی با FASTA نه تنها برای متخصصان بیوانفورماتیک، بلکه برای تمام محققان حوزههای زیستشناسی، ژنتیک و علوم زیستی مفید و ضروری است.
FASTA به عنوان یک فرمت فایل ساده اما قدرتمند و یک الگوریتم جستجوی توالی دقیق، نقش بنیادی در توسعه و پیشرفت علم بیوانفورماتیک داشته است. در طول بیش از سه دهه، FASTA علیرغم ظهور ابزارهای جدیدتر و سریعتر، همچنان جایگاه خود را به عنوان یکی از ارکان اصلی در تحلیل توالیهای بیولوژیکی حفظ کرده است. فرمت فایل FASTA با ساختار ساده و انعطافپذیر خود، استاندارد جهانی برای تبادل دادههای توالی است که تقریباً تمام نرمافزارها و دیتابیسهای بیوانفورماتیک از آن پشتیبانی میکنند.
الگوریتم FASTA با تمرکز بر دقت و قابلیت تنظیم، همچنان گزینه مناسبی برای کاربردهای خاص مانند شناسایی همولوگهای دور و تحلیلهای حساس توالی است. نسخههای جدیدتر این الگوریتم با بهرهگیری از فناوریهای پیشرفته مانند محاسبات موازی و پردازش ابری، کارایی و مقیاسپذیری بهتری را ارائه میدهند. کاربردهای متنوع FASTA از شناسایی توالیهای مشابه در مطالعات تکاملی، تا شناسایی گونه و پیشبینی عملکرد پروتئینها، بیانگر انعطافپذیری و اهمیت این ابزار در تحلیلهای زیستی است.
FASTA یک فرمت فایل متنی است که برای ذخیرهسازی توالیهای نوکلئوتیدی (DNA/RNA) یا پروتئینی استفاده میشود. همچنین نام الگوریتمی است که برای جستجوی شباهت بین توالیها کاربرد دارد.
در بیوانفورماتیک، «FASTA» میتواند هم به فایل فرمت متنی و هم به بستهٔ نرمافزاری جستوجوی توالی اشاره کند.
بستهٔ نرمافزاری FASTA که در EBI ارائه میشود، بهطور رسمی «fast A» (فَست اِی) تلفظ میشود و نشاندهندهٔ «FAST-All» است.
اما هنگام اشاره به فرمت فایل متنی FASTA، اغلب جامعهٔ کاربران آن را بهصورت «fast-ah» (فَست-آ) یا «fast-uh» (فَست-ا) ادا میکنند.
یک فایل FASTA شامل دو بخش اصلی است:
Header (خط توصیف): با علامت “<” شروع میشود و شامل شناسه یا توضیحات توالی است.
Sequence (توالی): خود توالی نوکلئوتیدی یا پروتئینی است که در خطوط بعدی قرار دارد.
FASTA فقط اطلاعات توالی را ذخیره میکند، در حالی که FASTQ علاوه بر توالی، دادههای کیفیت مربوط به هر نوکلئوتید را نیز ذخیره میکند. FASTQ برای دادههای حاصل از توالییابی نسل جدید (NGS) مناسبتر است.

تیم تولید محتوای گروه بیوانفورماتیک وانیار در تلاش است تا بهترین آموزشهای کوتاه در زمینه بیوانفورماتیک و زیستشناسی را تهیه نماید. صحت محتوای این صفحه توسط کارشناسان گروه بیوانفورماتیک وانیار بررسی شده است.



عضویت در مجله وانیار
جدید ترین مقالات در ایمیل شما!
با عضویت در مجله بیوانفورماتیک وانیار ، برترین مقالات را در ایمیل خود دریافت کنید.
سلام، وقت بخیر.
چطور میتونیم بهتون کمک کنیم؟ 🤓
تیم ما آماده پاسخگویی به سوالات شماست.
پشتیبانی 24 ساعته در 7 روز هفته.






