
FastQC در سال 2010 توسط Simon Andrews در موسسه بابراهام (Babraham Institute) توسعه یافت و از آن زمان به یکی از استانداردهای طلایی در ارزیابی کیفیت دادههای توالییابی تبدیل شده است. این ابزار رایگان و متنباز با هدف ارائه روشی ساده و سریع برای بررسی کیفیت دادههای FASTQ طراحی شده است. محبوبیت FastQC عمدتاً به دلیل رابط کاربری گرافیکی آسان، قابلیت اجرا از طریق خط فرمان (command line)، سرعت بالای پردازش و گزارشهای تصویری غنی است که امکان تفسیر سریع وضعیت دادهها را فراهم میکند. امروزه FastQC جزء جداییناپذیر اکثر پایپلاینهای تحلیل دیتای توالییابی نسل جدید (NGS) در حوزههای مختلف از جمله RNA-Seq، DNA-Seq، ChIP-Seq و میکروبیوم است.
FastQC ابزاری سبک و قابل نصب روی اغلب سیستمعاملهای رایج است که با حداقل پیشنیازها میتوان آن را بهسرعت راهاندازی کرد. این ابزار علاوه بر پشتیبانی از روشهای مختلف نصب در ویندوز، لینوکس و macOS، امکان اجرا هم از طریق رابط گرافیکی و هم خط فرمان را فراهم میکند و بهراحتی در تحلیلهای دستی یا پایپلاینهای اتوماتیک قابل استفاده است.
رمز فایل فشرده: www.vanyarbioinf.ir
FastQC مبتنی بر Java است و با Java 8 و نسخههای جدیدتر سازگار است؛ در نسخههای جدیدتر، استفاده از Java 11 یا بالاتر توصیه میشود. نصب این ابزار در پلتفرمهای مختلف به سادگی امکانپذیر است.
در سیستمعامل ویندوز، میتوانید فایل اجرایی را از وبسایت رسمی دانلود کرده و مستقیماً اجرا کنید.
در لینوکس، میتوانید از مدیر بسته (package manager) استفاده کنید، مثلا در توزیعهای مبتنی بر دبیان از دستور sudo apt-get install fastqc استفاده کنید یا فایل فشرده را دانلود و استخراج نمایید.
برای کاربران macOS، استفاده از Homebrew با دستور brew install fastqc راه سادهای برای نصب است.
همچنین، FastQC در بسیاری از مدیر بستههای بیوانفورماتیک مانند Bioconda نیز در دسترس است که با دستور conda install -c bioconda fastqc قابل نصب است.
FastQC را میتوان به دو روش اصلی اجرا کرد: رابط گرافیکی (GUI) و خط فرمان (CLI).
برای استفاده از رابط گرافیکی، کافی است فایل اجرایی را باز کنید و فایلهای FASTQ خود را از طریق منوی “File” بارگذاری کنید. این روش برای تحلیل تعداد کمی فایل مناسب است. برای تحلیل حجم بالای داده یا اتوماسیون، استفاده از خط فرمان کارامدتر است.
دستور پایه برای اجرای FastQC در خط فرمان به صورت زیر است:
fastqc filename.fastq.gzبرای پردازش همزمان چندین فایل میتوانید از دستور زیر استفاده کنید:
fastqc file1.fastq.gz file2.fastq.gz
OR
fastqc *.fastq.gz پارامترهای مفید دیگر شامل -o برای تعیین دایرکتوری خروجی، -t برای تعیین تعداد هستههای پردازشی و --extract برای استخراج خودکار فایلهای گزارش است.
FastQC کیفیت دیتای توالییابی را از زوایای مختلف و در قالب مجموعهای از ماژولهای مستقل بررسی میکند که هرکدام جنبهای خاص از کیفیت خوانشها (reads)، ترکیب بازها و آرتیفکتهای فنی را ارزیابی میکنند. تفسیر صحیح این ماژولها نیازمند در نظر گرفتن نوع کتابخانه، پلتفرم توالییابی و هدف تحلیل است، زیرا برخی الگوهای کیفی در انواع خاصی از دادههای NGS طبیعی محسوب میشوند. در ادامه تعدادی از این ماژولها را باهم بررسی میکنیم:
اولین ماژول FastQC، آمار پایه را ارائه میدهد که شامل نام فایل، نوع کدگذاری، تعداد کل توالیها، طول خوانشها و درصد GC است. این اطلاعات دید کلی از دادهها ارائه میدهد و میتواند در شناسایی مشکلات اولیه مانند تعداد غیرمنتظره خوانشها یا محتوای GC نامتعارف کمک کند. محتوای GC به طور خاص مهم است، زیرا انحراف قابل توجه از مقدار مورد انتظار برای ارگانیسم مورد مطالعه میتواند نشاندهنده آلودگی یا سوگیری در کتابخانه باشد. همچنین، این ماژول نوع کدگذاری نمرات کیفیت (Quality score encoding) را تشخیص میدهد. در دادههای مدرن Illumina، این کدگذاری مطابق استاندارد Sanger یا Phred-33 است و هر کاراکتر در خط چهارم FASTQ نشاندهنده کیفیت یک نوکلئوتید بر اساس این مقیاس است.
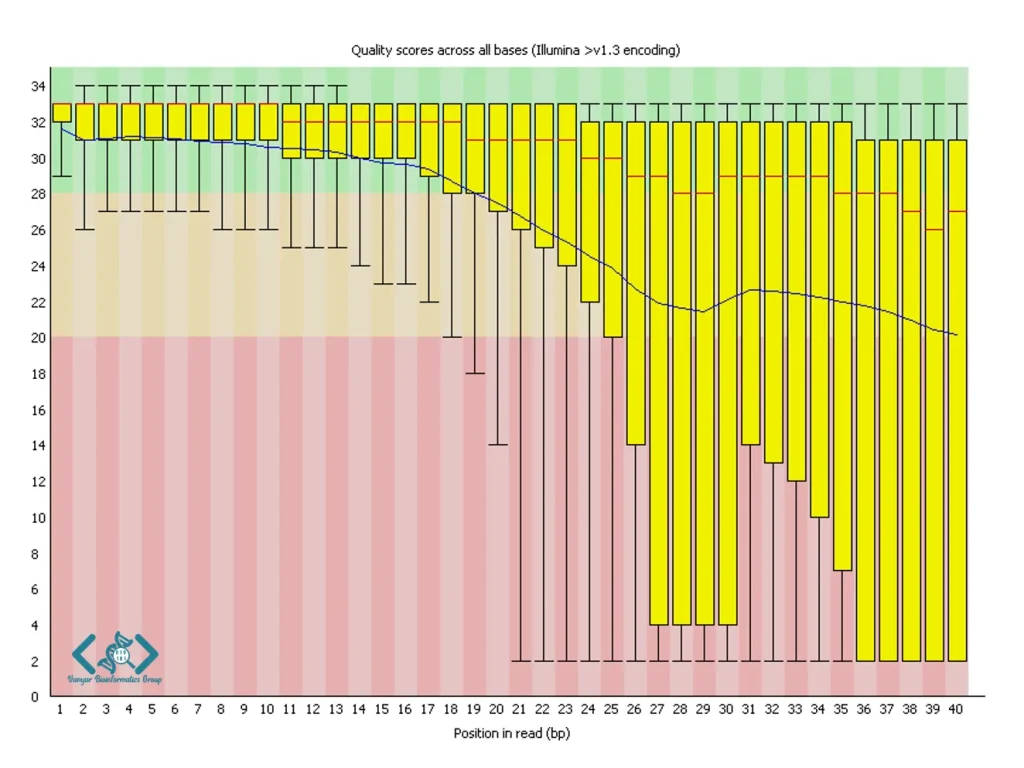
این ماژول یکی از مهمترین بخشهای گزارش FastQC است که نمودار جعبهای از توزیع نمرات کیفیت در هر موقعیت باز را نشان میدهد. محور افقی موقعیت بازها در طول خوانش و محور عمودی نمرات کیفیت (معمولاً در مقیاس Phred) را نمایش میدهد. به طور معمول، انتظار میرود کیفیت در ابتدای خوانش بالا باشد و به تدریج در انتهای خوانش کاهش یابد. بهطور تجربی، نمرات کیفیت بالاتر از Q28 معمولاً بسیار خوب، Q20–28 قابل قبول و زیر Q20 پایین در نظر گرفته میشوند؛ با این حال، FastQC بهصورت رسمی آستانه عددی ثابت تعریف نمیکند و ارزیابی بر اساس توزیع داده انجام میشود. افت شدید کیفیت در انتهای خوانشها پدیدهای رایج است که میتواند با تریمینگ اصلاح شود. همچنین، الگوهای غیرمعمول مانند افت ناگهانی کیفیت در موقعیتهای خاص میتواند نشاندهنده مشکلات فنی در فرآیند توالییابی باشد.
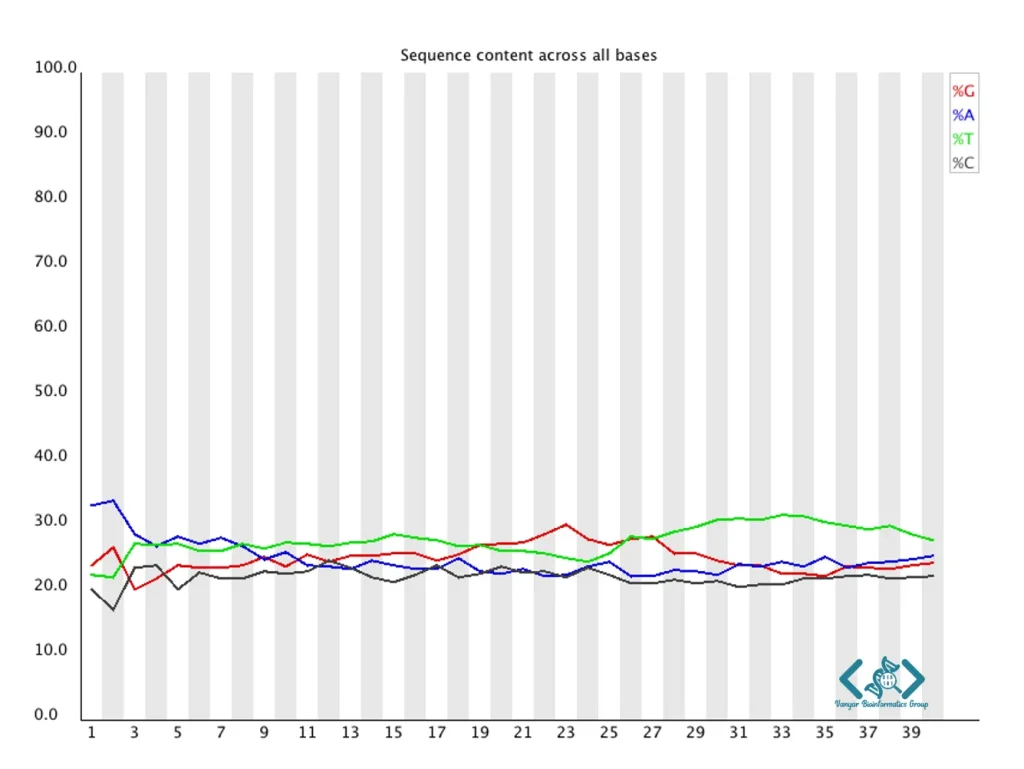
این ماژول نسبت چهار نوکلئوتید (A، T، G، C) را در هر موقعیت خوانش نشان میدهد. در یک کتابخانه متوازن و بدون سوگیری با fragmentation تصادفی (مانند توالییابی کل اگزوم یا WES)، انتظار میرود نسبت نوکلئوتیدها در هر موقعیت تقریباً ثابت باشد، با تفاوتهای کوچکی که بازتابدهنده محتوای ژنومی است. تفاوتهای قابل توجه در محتوای بازها، بهویژه در ابتدای خوانشها، معمولاً نشاندهنده سوگیری در فرآیند کتابخانهسازی است. این سوگیری در برخی پروتکلها مانند RNA-Seq با پرایمرهای هگزامر تصادفی طبیعی است. با این حال، در DNA-Seq، انحرافهای شدید میتواند نشاندهنده مشکلاتی مانند آداپتورهای باقیمانده یا آلودگی باشد.
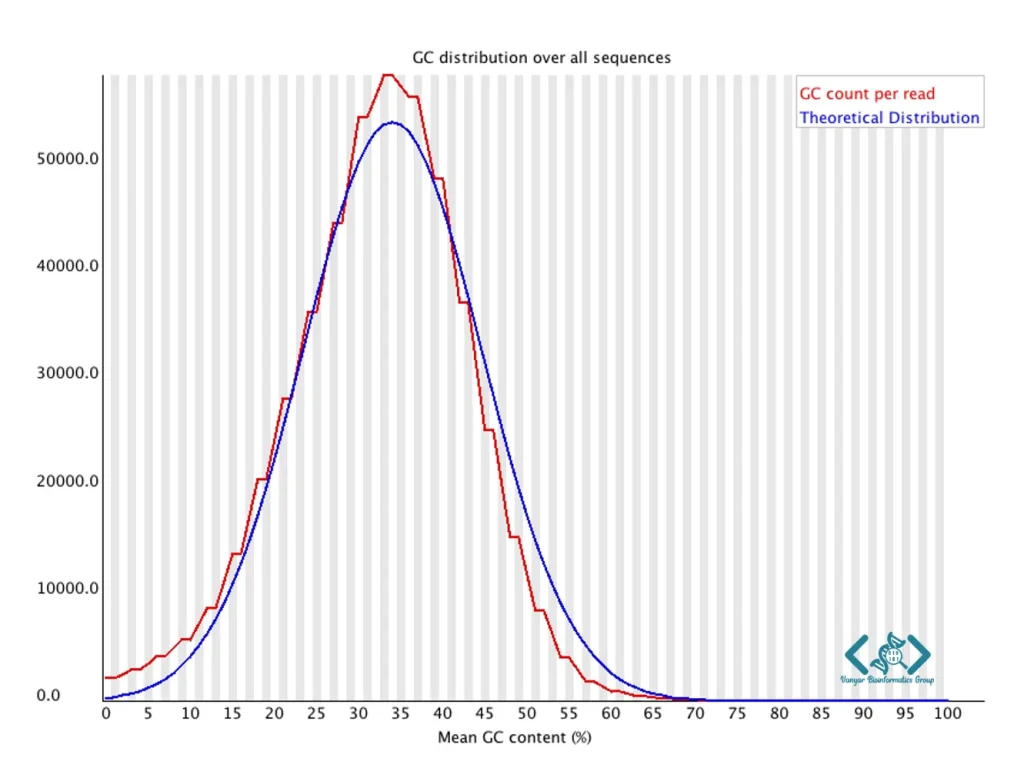
این ماژول توزیع محتوای GC را در کل خوانشها نشان میدهد و آن را با توزیع نظری مورد انتظار مقایسه میکند. در کتابخانههای تصادفی، توزیع GC نزدیک به مدل نظری نشانه کیفیت مناسب است؛ اما در کتابخانههای هدفمند یا RNA-Seq، انحراف از این الگو لزوماً نشاندهنده مشکل نیست. انحراف از این الگو، مانند توزیع دو قلهای یا شانهدار، میتواند نشاندهنده آلودگی با ارگانیسمهای دیگر، سوگیری در تکثیر PCR یا مشکلات در فرآیند کتابخانهسازی باشد. برای مثال، حضور یک قله ثانویه با محتوای GC متفاوت اغلب نشاندهنده آلودگی باکتریایی در نمونههای یوکاریوتی است. این ماژول بهویژه برای شناسایی آلودگیهای میکروبی یا مخلوطهای نمونهای مفید است.
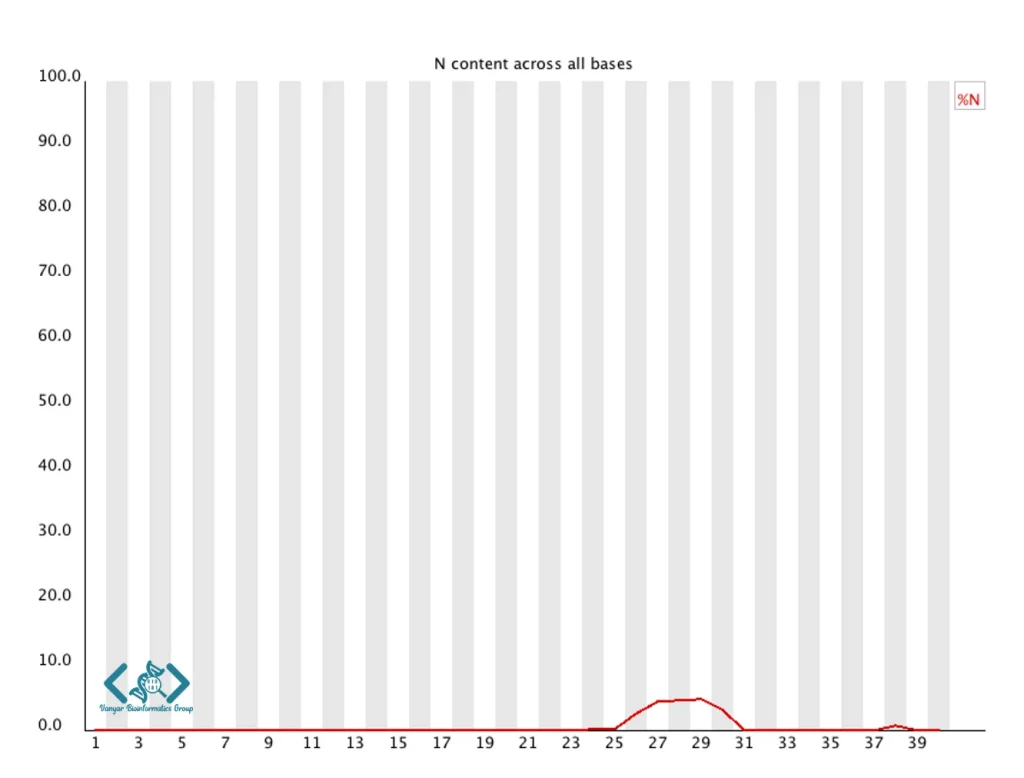
این ماژول درصد نوکلئوتیدهای نامشخص (N) را در هر موقعیت خوانش نمایش میدهد. در دادههای با کیفیت بالا، انتظار میرود محتوای N نزدیک به صفر باشد. افزایش محتوای N معمولاً نشاندهنده مشکلات فنی در فرایند توالییابی است. بهطور خاص، افزایش محتوای N در انتهای خوانشها میتواند نشاندهنده افت کیفیت سیگنال باشد، در حالیکه الگوهای منظم یا افزایش ناگهانی در موقعیتهای خاص ممکن است نشاندهنده مشکلات دستگاهی باشد. سطوح بالای N میتواند تحلیلهای پاییندستی مانند الاینمنت و فراخوانی واریانت (variant calling) را تحت تأثیر قرار دهد، بنابراین خوانشهای با محتوای N بالا معمولاً باید فیلتر یا هرس (Trim) شوند.
این ماژول حضور توالیهای آداپتور را در دادهها بررسی میکند. آداپتورها توالیهای مصنوعی هستند که در فرایند کتابخانهسازی به قطعات DNA متصل میشوند. در خوانشهای طولانیتر از قطعات DNA اصلی، بخشی از آداپتور نیز خوانده میشود. FastQC مجموعهای از توالیهای آداپتور رایج را جستجو کرده و درصد خوانشهایی که حاوی این توالیها هستند را در هر موقعیت گزارش میکند. حضور آداپتور، بهویژه در انتهای خوانشها، رایج است و باید قبل از تحلیلهای پاییندستی با استفاده از ابزارهای تریمینگ مانند Trimmomatic یا Cutadapt حذف شوند.
گزارشهای FastQC تنها زمانی معنا پیدا میکنند که در چارچوب نوع کتابخانه، پلتفرم توالییابی و هدف تحلیل تفسیر شوند. شناسایی درست مشکلات کیفی و انتخاب راهکارهای پیشپردازش مناسب، نقش کلیدی در بهبود کیفیت دیتا و افزایش دقت تحلیلهای پاییندستی دارد.
FastQC نتایج هر ماژول را با سه رنگ سبز (pass)، نارنجی (warn) و قرمز (fail) نشان میدهد. با این حال، تفسیر این نتایج نیازمند درک زمینه بیولوژیکی و نوع کتابخانه است. برخی از مشکلات رایج شامل: کیفیت پایین در انتهای خوانشها، آلودگی آداپتور، سوگیری یا بایاس در محتوای باز، سطوح بالای تکرار (duplication) و توزیع غیرطبیعی GC است.
مهم است بدانیم که برخی هشدارها در انواع خاصی از کتابخانهها طبیعی است. برای مثال، در RNA-Seq سطوح بالای تکرار به دلیل تفاوت در سطح بیان ژنها قابل انتظار است. تفسیر صحیح نتایج FastQC نیازمند در نظر گرفتن این زمینههای بیولوژیکی و فنی است.
راهکارهای پیشپردازش دیتا بر اساس گزارش FastQC
پس از شناسایی مشکلات کیفی، اقدامات اصلاحی مناسب باید انجام شود.
🟠 تریمینگ کیفیت با استفاده از ابزارهایی مانند Trimmomatic، BBDuk یا fastp میتواند بازهای با کیفیت پایین در انتهای خوانشها را حذف کند.
🔴 برای آلودگی آداپتور، ابزارهایی مانند Cutadapt یا Trimmomatic با پارامترهای ILLUMINACLIP میتوانند توالیهای آداپتور را شناسایی و حذف کنند.
🟣 فیلترینگ خوانشها بر اساس طول، کیفیت میانگین یا محتوای N نیز میتواند کیفیت کلی مجموعه داده را بهبود بخشد.
⚠️ در موارد سوگیری شدید در محتوای GC یا آلودگی، ممکن است نیاز به تکرار آزمایش باشد.
پس از پیشپردازش، اجرای مجدد FastQC برای تأیید بهبود کیفیت توصیه میشود. استراتژی پیشپردازش باید متناسب با نوع کتابخانه و تحلیلهای پاییندستی تنظیم شود، زیرا برخی تحلیلها به کیفیت بالاتر نیاز دارند.
در این بخش میبینید که FastQC چگونه با توجه به نوع دیتای NGS رفتارهای متفاوتی را آشکار میکند و چه نکاتی را باید در هر پلتفرم توالییابی زیر نظر داشت. با مرور این کاربردها متوجه میشوید که تفسیر خروجی FastQC تنها یک چک ساده نیست، بلکه نیازمند درک نوع داده و انتظارات بیولوژیکی آن است.
FastQC در تحلیل RNA-Seq کمک میکند تا مشکلات خاص این نوع دیتا را شناسایی کنیم. به عنوان مثال، سوگیری در محتوای باز در ابتدای خوانشها در RNA-Seq با پرایمرهای هگزامر تصادفی طبیعی است. همچنین، سطوح بالای تکرار به دلیل بیان متفاوت ژنها (برخی ژنها بسیار پربیان هستند) قابل انتظار است و لزوماً نشاندهنده مشکل فنی نیست.
در RNA-Seq، توجه خاص به کیفیت کلی خوانشها و حذف آداپتورها اهمیت دارد، زیرا این عوامل میتوانند بر دقت تخمین بیان ژن تأثیر بگذارند. برای RNA-Seq با خوانشهای جفتی (paired-end)، بررسی کیفیت هر دو فایل FASTQ ضروری است. در برخی موارد، تریمینگ شدیدتر برای دادههای RNA-Seq نسبت به DNA-Seq توصیه میشود، زیرا دقت الاینمنت خوانشها به اگزونها و شناسایی محلهای پیرایش (splice sites) اهمیت بیشتری دارد.
FastQC در توالییابی DNA به شناسایی مشکلاتی مانند آلودگی، سوگیری در تکثیر و کیفیت پایین کمک میکند. برخلاف RNA-Seq، در DNA-Seq انتظار میرود محتوای باز در تمام موقعیتها نسبتاً متوازن باشد و سطوح تکرار پایین باشد. در ChIP-Seq، به دلیل غنیسازی یا هدفگیری مناطق خاص، سطوح بالاتر تکرار طبیعی است. در هر دو نوع دیتا، حضور آداپتور، بهویژه در خوانشهای کوتاه، میتواند بر دقت الاینمنت و فراخوانی واریانت تأثیر منفی بگذارد. کیفیت بالا در DNA-Seq برای تشخیص واریانتها اهمیت ویژهای دارد، زیرا خطاهای توالییابی میتوانند به اشتباه به عنوان واریانت تفسیر شوند.
در ChIP-Seq، انتظار میرود خوانشها بهصورت غیریکنواخت و در قالب نواحی غنیشده (peaks) در ژنوم مرجع تجمع پیدا کنند. با این حال، سوگیریهای فنی مانند سوگیری GC میتوانند منجر به شناسایی نادرست پیکها شوند و باید در تفسیر دادهها و مراحل پاییندستی تحلیل مورد توجه قرار گیرند.
در مطالعات میکروبیوم و متاژنومیک، FastQC نقش مهمی در شناسایی آلودگیها و ارزیابی تنوع نمونه دارد. در این نوع دیتا، توزیع محتوای GC معمولاً چندقلهای است که بازتابدهنده تنوع گونههای میکروبی با محتوای GC متفاوت است. بنابراین، هشدار در ماژول محتوای GC لزوماً نشاندهنده مشکل نیست. با این حال، قلههای غیرمنتظره میتواند نشاندهنده آلودگی با منابع غیرمیکروبی باشد. در این پروژهها، توجه ویژه به کیفیت خوانشها اهمیت دارد، زیرا خطاهای توالییابی میتوانند به تخمین نادرست تنوع میکروبی منجر شوند. همچنین، شناسایی و حذف توالیهای میزبان (مثلاً انسان در مطالعات میکروبیوم روده) با استفاده از ابزارهای تکمیلی مهم است که FastQC میتواند در شناسایی اولیه این آلودگیها کمک کند.
MultiQC ابزاری قدرتمند برای تجمیع نتایج FastQC از چندین نمونه در یک گزارش واحد است. این ابزار گزارشهای HTML تعاملی تولید میکند که امکان مقایسه سریع معیارهای کیفی بین نمونهها را فراهم میکند.
برای استفاده از MultiQC، کافی است دستور multiqc fastqc_results/ را اجرا کنید تا تمام گزارشهای FastQC در دایرکتوری مشخص شده تجمیع شوند.
⚓ MultiQC علاوه بر FastQC، نتایج بسیاری از ابزارهای دیگر مانند Trimmomatic، STAR، Bowtie2 و Picard را نیز تجمیع میکند، بنابراین میتواند برای ایجاد گزارشهای جامع از کل فرآیند تحلیل استفاده شود. این قابلیت بهویژه در پروژههای بزرگ با تعداد زیاد نمونه مفید است و به شناسایی سریع نمونههای مشکلدار یا روندهای کلی در کیفیت دادهها کمک میکند.
با وجود کاربرد گسترده FastQC در ارزیابی کیفیت دادههای NGS، این ابزار همهی جنبههای کیفی و همهی انواع دیتای توالییابی را پوشش نمیدهد. شناخت محدودیتهای FastQC و استفاده از ابزارهای تکمیلی متناسب با نوع دیتا و هدف تحلیل، برای دستیابی به ارزیابی کیفی دقیقتر و قابلاعتماد ضروری است.
با وجود کاربردهای گسترده، FastQC محدودیتهایی نیز دارد:
🟣 این ابزار برای دادههای توالییابی با خوانش کوتاه بهینه شده و ممکن است برای تحلیل دیتای توالییابی طولانی مانند PacBio یا Oxford Nanopore مناسب نباشد.
🔵 همچنین، FastQC توالیهای آداپتور محدودی را به صورت پیشفرض جستجو میکند و ممکن است آداپتورهای سفارشی یا کمتر رایج را تشخیص ندهد.
🟢 در دادههای خاص مانند تکسلولی (single-cell) یا کتابخانههای با سوگیری ذاتی (مانند ChIP-Seq یا ATAC-Seq)، هشدارهای FastQC ممکن است گمراهکننده باشد.
🔴 این ابزار همچنین قادر به تشخیص برخی مشکلات پیچیده مانند آلودگی متقاطع بین نمونهها یا خطاهای دیمولتیپلکسینگ نیست.
برای غلبه بر این محدودیتها، استفاده از ابزارهای تکمیلی و تفسیر نتایج با در نظر گرفتن زمینه بیولوژیکی و فنی ضروری است.
برای کنترل کیفیت جامعتر، FastQC میتواند با ابزارهای دیگر تکمیل شود. FastqScreen برای شناسایی آلودگیهای ژنومی از منابع مختلف مفید است. Kraken2 یا Centrifuge میتوانند برای شناسایی دقیقتر آلودگیهای میکروبی استفاده شوند. برای دادههای توالییابی طولانی، ابزارهایی مانند NanoPlot یا LongQC طراحی شدهاند. Picard CollectSequencingArtifactMetrics برای شناسایی سوگیریهای اکسیداتیو و سایر آرتیفکتهای شیمیایی مفید است. FASTK و Jellyfish برای تحلیل دقیقتر k-mer و شناسایی تکرارها و آلودگیها کاربرد دارند. همچنین، ابزارهای تخصصی برای انواع خاص داده مانند RNA-QC-Chain برای RNA-Seq یا ATACseqQC برای دادههای ATAC-Seq وجود دارند. ترکیب این ابزارها با FastQC میتواند دید جامعتری از کیفیت دادهها ارائه دهد و به شناسایی مشکلاتی که FastQC به تنهایی قادر به تشخیص آنها نیست، کمک کند.
با افزایش حجم و پیچیدگی دادههای توالییابی، روشهای کنترل کیفیت نیز در حال تکامل هستند.
🔹یکی از روندهای مهم، توسعه ابزارهای کنترل کیفیت با قابلیت یادگیری ماشین است که میتوانند الگوهای پیچیده خطا را شناسایی کنند و با انواع مختلف دیتا سازگار شوند.
🔹همچنین، ابزارهای کنترل کیفیت در زمان واقعی (real-time) در حال توسعه هستند که میتوانند دادهها را حین تولید ارزیابی کرده و بازخورد فوری ارائه دهند.
🔹ادغام اطلاعات کنترل کیفیت با متادیتای نمونه و دادههای آزمایشگاهی نیز روندی رو به رشد است که امکان شناسایی منابع خطا در مراحل قبل از توالییابی را فراهم میکند.
🔹علاوه بر این، با افزایش محبوبیت توالییابی تکسلولی و فضایی، ابزارهای کنترل کیفیت تخصصی برای این فناوریها در حال توسعه هستند که میتوانند معیارهای خاص مانند تعداد سلولها، نرخ doublet و کیفیت بارکد را ارزیابی کنند.
FastQC یکی از ابزارهای کلیدی در کنترل کیفیت دادههای توالییابی است که امکان ارزیابی سریع و قابلاعتماد کیفیت دادههای NGS را فراهم میکند. گزارشهای تصویری و قابلتفسیر این ابزار به شناسایی مشکلات کیفی و انتخاب راهکارهای اصلاحی مناسب کمک میکند. با وجود برخی محدودیتها، FastQC به دلیل سادگی، سرعت و کارایی، همچنان جایگاه ویژهای در آنالیز دادههای توالییابی دارد. ادغام آسان FastQC در پایپلاینهای خودکار NGS، آن را به نخستین گام استاندارد در بسیاری از تحلیلها تبدیل کرده است. در نهایت، تفسیر صحیح نتایج FastQC در کنار سایر ابزارهای تکمیلی، نقش مهمی در تضمین دقت، اعتبار و تکرارپذیری نتایج علمی ایفا میکند.
FastQC یک ابزار کنترل کیفیت برای فایلهای FASTQ است که مشکلاتی مثل افت کیفیت، آداپتور و ناهنجاریهای GC را نشان میدهد.
بله، FastQC بهصورت رایگان در دسترس کاربران قرار دارد و میتوانید گزارشهای کنترل کیفیت را بدون هزینه تهیه کنید.
بله؛ FastQC برای ویندوز، لینوکس و macOS با روشهای مختلف (GUI/CLI و بستههای مدیریتی) قابل نصب است.
FastQC برای دادههای خوانش کوتاه (short-read) بهینه است و برای برخی پلتفرمهای long-read مثل PacBio/Oxford Nanopore ممکن است دقیق نباشد.
یکی از مهمترین کاربردهای FastQC تشخیص آداپتورهای باقیمانده در انتهای خوانشهاست (که اگر اصلاح نشود نتایج تحلیل میتواند دچار خطا شود).

تیم تولید محتوای گروه بیوانفورماتیک وانیار در تلاش است تا بهترین آموزشهای کوتاه در زمینه بیوانفورماتیک و زیستشناسی را تهیه نماید. صحت محتوای این صفحه توسط کارشناسان گروه بیوانفورماتیک وانیار بررسی شده است.




عضویت در مجله وانیار
چطور از جدیدترین آموزشها باخبر شوم؟
با عضویت در مجله بیوانفورماتیک وانیار، برترین آموزشهای بیوانفورماتیک را در لحظه انتشار دریافت کنید.
سلام، وقت بخیر.
چطور میتونیم بهتون کمک کنیم؟
تیم ما آماده پاسخگویی به سوالات شماست.
پشتیبانی 24 ساعته در 7 روز هفته.


