
در دنیای پیچیده توالییابی نسل جدید (NGS)، کیفیت دیتای خام نقش تعیینکنندهای در اعتبار نتایج نهایی دارد. Trimmomatic یکی از ابزارهای قدرتمند و پرکاربرد در حوزه پیشپردازش (preprocessing) دادههای توالییابی شرکت Illumina است که در «آزمایشگاه اوسادل» (Usadel Lab) توسعه یافته است. این ابزار با هدف بهبود کیفیت دیتای خام توالییابی از طریق حذف توالیهای آداپتور، برش نواحی با کیفیت پایین و فیلتر کردن خوانشهای (reads) کوتاه طراحی شده است.
Trimmomatic قابلیت پردازش دیتای تکخوانش (single-end) و جفتخوانش (paired-end) را دارد و به عنوان یک نرمافزار مبتنی بر جاوا، در اکثر سیستمعاملها قابل اجراست.
در حالیکه بدنه اصلی نرمافزار تحت مجوز GPL منتشر شده، توالیهای آداپتور موجود در آن مشمول این مجوز نمیشوند. مالکیت این توالیها متعلق به شرکت Illumina است و با کسب اجازه از آنها در این ابزار استفاده شدهاند.
در این بخش، به اهمیت پیشپردازش دادههای توالییابی نسل جدید و چالشهای کیفی رایج در این نوع دیتا میپردازیم؛ سپس نقش ابزار Trimmomatic را در رفع این مشکلات و بهبود کیفیت دادهها تشریح میکنیم.
دادههای خام حاصل از توالییابی نسل جدید معمولاً حاوی خطاها و نویزهایی هستند که میتوانند نتایج تحلیلهای بعدی را تحت تأثیر قرار دهند. این خطاها شامل توالیهای آداپتور، نوکلئوتیدهای بیکیفیت در انتهای خوانشها و آلودگیهای احتمالی هستند. پیشپردازش دادهها گامی حیاتی برای اطمینان از صحت نتایج در فرایندهایی مانند همترازی (Alignment) خوانشها، سرهم کردن ژنوم (Genome Assembly)، شناسایی واریانتها (Variant Calling) و آنالیز بیان ژن محسوب میشود.
بدون پیشپردازش مناسب، حضور توالیهای آداپتور میتواند فرایند همترازی خوانشها با ژنوم مرجع را مختل کند، در حالیکه نواحی با کیفیت پایین میتوانند به شناسایی واریانتهای کاذب بینجامند. Trimmomatic با حذف این منابع خطا، کیفیت کلی دیتا را بهبود میبخشد و اعتبار نتایج نهایی را افزایش میدهد. مطالعات مختلف نشان دادهاند که پیشپردازش مناسب با ابزارهایی مانند Trimmomatic میتواند باعث بهبود معنیدار در نرخ الاینمنت خوانشها، کاهش خطاهای واریانتیابی و افزایش اطمینان از نتایج آنالیزهای پاییندستی شود.
پلتفرمهای مختلف توالییابی نسل جدید، هر کدام چالشهای خاص خود را در زمینه کیفیت دیتا دارند.
برای مثال، دادههای Illumina معمولاً با افت کیفیت در انتهای ریدها مواجه هستند، در حالی که پلتفرمهای Ion Torrent چالشهایی در توالیهای هموپلیمر دارند. علاوه بر این، طول و ترکیب آداپتورها در پروتکلهای مختلف متفاوت است و شناسایی و حذف آنها نیازمند ابزاری انعطافپذیر مانند Trimmomatic است.
فاکتورهای دیگری مانند کیفیت نمونه اولیه، روشهای آمادهسازی کتابخانه و خطاهای سیستماتیک دستگاههای توالییاب نیز بر کیفیت دادههای خام تأثیرگذارند. Trimmomatic با ارائه مجموعهای از فیلترها و پارامترهای قابل تنظیم، امکان مقابله با این چالشها را فراهم میکند و به محققان اجازه میدهد تا بر اساس نوع دیتا و هدف پروژه، استراتژی مناسب پیشپردازش را اتخاذ کنند.
در ادامه، به معرفی پیشنیازهای نرمافزار Trimmomatic میپردازیم و روشهای مختلف نصب آن را بههمراه نحوه بررسی صحت نصب، آموزش میدهیم.
Trimmomatic یک ابزار Cross-Platform است که برای اجرا به Java Runtime Environment نیاز دارد. نصب بودن Java بر روی سیستم شما، اجرای Trimmomatic را بر روی هر سیستمعاملی ممکن میسازد.
دو روش اصلی برای راهاندازی این نرمافزار، شامل دانلود مستقیم فایل اجرایی و نصب از طریق مدیریت پکیج conda، توضیح داده شده است:
در این روش، میتوانید فایل فشرده Trimmomatic را مستقیماً از این لینک زیر دانلود کنید:
رمز فایل فشرده: www.vanyarbioinf.ir
یا نرمافزار را با کد زیر، از لینک وبسایت رسمی نرمافزار دانلود کنید:
wget http://www.usadellab.org/cms/uploads/supplementary/Trimmomatic/Trimmomatic-0.39.zip
پس از دریافت فایل، کافی است آن را از حالت فشرده خارج کرده و وارد پوشه برنامه شوید:
unzip Trimmomatic-0.39.zip
cd Trimmomatic-0.39فایل اجرایی اصلی با نام trimmomatic-0.39.jar در این پوشه قرار دارد.
برای کاربرانی که از مدیریت پکیج conda استفاده میکنند، Trimmomatic از طریق کانالهای Bioconda نیز قابل نصب است:
conda install -c bioconda trimmomaticدر این روش، دستور trimmomatic بهصورت مستقیم در ترمینال در دسترس خواهد بود و نیازی به اجرای فایل JAR نیست.
برای بررسی صحت نصب در روش دانلود مستقیم، میتوان دستور زیر را اجرا کرد:
java -jar trimmomatic-0.39.jarاگر نصب بهدرستی انجام شده باشد، صفحه راهنمای Trimmomatic بههمراه شماره نسخه نمایش داده میشود.
در روش نصب با Conda، دستور زیر کافی است:
trimmomaticدر صورت بروز خطا، باید از نصب صحیح Java و صحت مسیر فایل اجرایی Trimmomatic اطمینان حاصل کرد.
ابزار Trimmomatic دارای مجموعهای از پارامترهای قدرتمند است که امکان کنترل دقیق فرایند پیشپردازش دادههای NGS را فراهم میکنند. این پارامترها به طور کلی به دو دسته اصلی تقسیم میشوند:
-threads) است.در ادامه، مهمترین پارامترهای پردازشی و نحوه عملکرد آنها به ترتیب اولویت و استاندارد استفاده، تشریح شدهاند:
این مهمترین و معمولاً اولین فیلتر در خط فرمان است. وظیفه آن شناسایی و حذف توالیهای آداپتور و پرایمرهایی است که در طول توالییابی به خوانشها متصل شدهاند.
برای دادههای جفتخوانش (Paired-End)، فرمت کامل و استاندارد به صورت زیر است:
ILLUMINACLIP:<fastaWithAdapters>:<seed mismatches>:<palindrome clip threshold>:<simple clip threshold>:<minAdapterLength>:<keepBothReads>true یا false).این فیلتر یک پنجره متحرک را روی توالی حرکت میدهد و میانگین کیفیت بازها را در آن پنجره محاسبه میکند. اگر میانگین کیفیت در پنجره از آستانه تعیینشده کمتر شود، خوانش از آن نقطه به بعد بریده میشود و مابقی آن دور ریخته میشود.
SLIDINGWINDOW:<windowSize>:<requiredQuality>SLIDINGWINDOW:4:20 یعنی پنجرههای ۴ تایی را بررسی کن و هرجا میانگین کیفیت زیر ۲۰ شد، بقیه توالی را از آن نقطه حذف کن.این دو فیلتر به صورت مستقیم بازهای بیکیفیت را صرفاً از ابتدا (LEADING) یا انتهای (TRAILING) خوانش حذف میکنند تا زمانی که به یک باز با کیفیت مناسب برسند.
LEADING:<quality>
TRAILING:<quality>مثال: LEADING:3 یعنی از ابتدای خوانش شروع کن و هر بازی که کیفیتش زیر ۳ است را حذف کن تا به اولین باز با کیفیت بالای ۳ برسی.
این یک فیلتر پیشرفته و تطبیقی است که بر اساس طول خوانش و کیفیت آن تصمیمگیری میکند تا تعادلی بین “حفظ طول حداکثری” و “حذف خطاهای احتمالی” ایجاد کند.
MAXINFO:<targetLength>:<strictness>این فیلترها بدون توجه به کیفیت، تغییرات طولی اعمال میکنند و معمولاً برای حذف نواحی خاصی که مشکلات تکنیکال دارند استفاده میشوند:
CROP:<length>
HEADCROP:<length>CROP:150 یعنی فقط ۱۵۰ باز اول را نگه دار).HEADCROP:10 یعنی ۱۰ باز اول را دور بریز).این فیلتر معمولاً آخرین مرحله است و خوانشهایی را که پس از تمام مراحل بالا (حذف آداپتور و برش کیفیت) بیش از حد کوتاه شدهاند، به طور کامل حذف میکند تا وارد مراحل بعدی آنالیز (مانند Mapping) نشوند.
MINLEN:<length>مثال: MINLEN:36 یعنی اگر طول نهایی خوانش کمتر از ۳۶ باز بود، آن را کلاً دور بریز.
در این بخش، نمونه دستورات استاندارد را برای دو سناریوی رایج (تکخوانش و جفتخوانش) ارائه میکنیم:
در این دستور، Trimmomatic در حالت SE فراخوانی میشود. ترتیب فایلها به صورت «یک ورودی» و «یک خروجی» است.
java -jar trimmomatic-0.39.jar SE -threads 4 \
input.fastq.gz output.fastq.gz \
ILLUMINACLIP:TruSeq3-SE.fa:2:30:10 \
LEADING:3 TRAILING:3 \
SLIDINGWINDOW:4:15 \
MINLEN:36SE: حالت اجرا تکخوانش
threads 4: استفاده از ۴ هسته پردازشی
ILLUMINACLIP: حذف آداپتورهای TruSeq3 (مخصوص SE) با حداکثر ۲ عدم تطابق در Seed، و آستانههای برش ۳۰ و ۱۰
SLIDINGWINDOW:4:15: پنجره ۴ تایی با کیفیت میانگین ۱۵
MINLEN:36: حذف خوانشهای نهایی کوتاهتر از ۳۶ باز
در یک آزمایش مقایسهای (benchmarking)، نرمافزار Trimmomatic بر روی مجموعهای متشکل از 19 میلیون جفت Read (paired-end) با طول 150 جفتباز که توسط دستگاه Illumina تولید شده بود، با بهرهگیری از 8 رشته پردازشی (threads) مورد ارزیابی قرار گرفت. نتایج حاصل از این ارزیابی به شرح زیر بود:
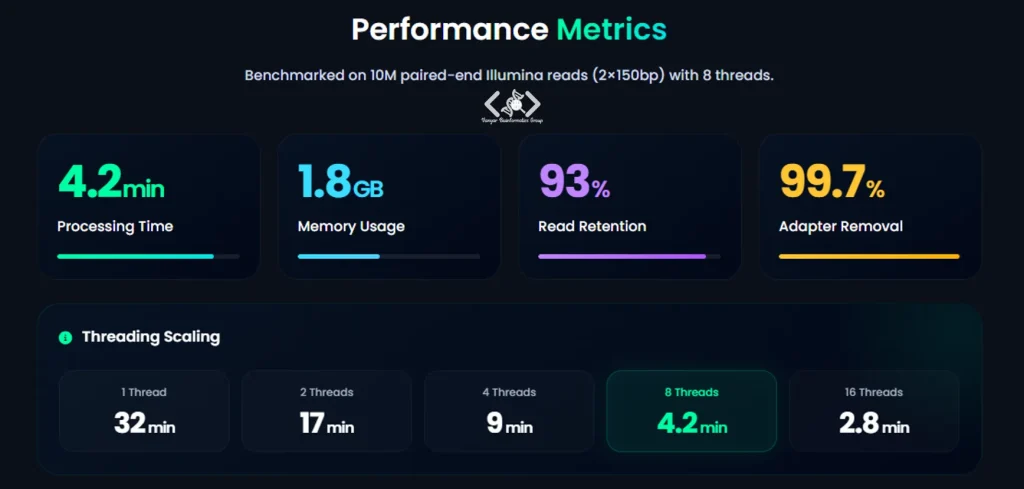
این معیارها نشان میدهد که Trimmomatic ابزاری کارآمد، سریع و دقیق برای پیشپردازش دادههای توالییابی نسل جدید، بهویژه دیتای paired-end، محسوب میشود.
در حالت PE، ترتیب فایلها بسیار حساس است: ابتدا دو فایل ورودی (رفت و برگشت)، سپس چهار فایل خروجی (به ترتیب: رفتِ جفتشده، رفتِ جفتنشده، برگشتِ جفتشده، برگشتِ جفتنشده).
java -jar trimmomatic-0.39.jar PE -threads 8 \
input_1.fastq.gz input_2.fastq.gz \
output_1P.fastq.gz output_1U.fastq.gz \
output_2P.fastq.gz output_2U.fastq.gz \
ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 \
LEADING:20 TRAILING:20 \
SLIDINGWINDOW:4:20 \
MINLEN:70output_1P و output_2P: حاوی خوانشهایی که هر دو جفت کیفیت لازم را داشتهاند (Paired)output_1U و output_2U: حاوی خوانشهایی که جفتشان حذف شده است (Unpaired)انتخاب ابزار مناسب برای پیشپردازش دادههای NGS، به تعادلی میان «دقت»، «سرعت» و «سهولت استفاده» نیاز دارد. در این بخش، جایگاه Trimmomatic را با سه رقیب اصلیاش بررسی میکنیم.
ابزار Cutadapt (نوشتهشده با Python) یکی از منعطفترین ابزارهای موجود است.
ابزار Trim Galore در واقع یک “Wrapper” (پوسته) است که ابزارهای Cutadapt و FastQC را ترکیب میکند.
ابزار BBDuk (توسعهیافته توسط JGI) رقیبی قدرتمند و مبتنی بر جاوا است.
در این بخش، به بررسی ویژگیهای کلیدی Trimmomatic میپردازیم و با مقایسه نقاط قوت آن در پردازش دیتای Illumina و محدودیتهای فنیاش در مقایسه با ابزارهای مدرن، دیدگاه مناسبی برای انتخاب آگاهانه این نرمافزار ارائه میکنیم.
ابزار Trimmomatic به عنوان یکی از ستونهای اصلی در فرایند آنالیز دادههای توالییابی (NGS)، نقش حیاتی در تبدیل دادههای خام و پرنویز به اطلاعاتی قابلاعتماد ایفا میکند. این نرمافزار با ارائه مجموعهای کامل از فیلترها—از حذف دقیق آداپتورها در دادههای Illumina گرفته تا برش هوشمند نواحی بیکیفیت با روش پنجره لغزان—تعادلی منطقی میان «حفظ دیتای مفید» و «حذف خطاهای سیستمی» برقرار میکند. اگرچه در مقایسه با ابزارهای مدرنتر، رابط کاربری آن کمی پیچیدهتر است، اما دقت بالای آن در پردازش دادههای جفتخوانش (Paired-end) و وجود منابع آموزشی گسترده، آن را به انتخابی مطمئن برای پژوهشگران تبدیل کرده است. در نهایت، استفاده صحیح از Trimmomatic تضمین میکند که نتایج حساس تحقیقاتی، مانند شناسایی واریانتها یا بررسی بیان ژن، بر پایه دادههایی سالم و معتبر استوار باشند.
Trimmomatic یک ابزار تخصصی در بیوانفورماتیک است که برای پیشپردازش و پاکسازی دادههای توالییابی نسل جدید (NGS) طراحی شده است. این نرمافزار با شناسایی و حذف آداپتورهای باقیمانده و برش (Trim) بخشهای دارای کیفیت پایین در انتهای خوانشها (Reads)، دقت نتایج نهایی را در تحلیلهای بعدی مانند الاینمنت و آنالیزهای ژنومی بهطور چشمگیری افزایش میدهد.
خیر، این نرمافزار کاملاً رایگان و متنباز (Open Source) است. شما میتوانید آخرین نسخه آن را بدون پرداخت هیچ هزینهای از وبسایت سازنده یا گیتهاب (GitHub) دانلود کرده و در پروژههای تحقیقاتی خود استفاده کنید.
دادههای خام حاوی آداپتورها و توالیهای بیکیفیتی هستند که باعث خطاهای سیستماتیک در مراحل بعدی (مانند الاینمنت و فراخوانی واریانتها) میشوند. Trimmomatic با حذف این نویزها، از بروز نتایج گمراهکننده و واریانتهای کاذب جلوگیری کرده و دقت تحلیلهای بیوانفورماتیکی شما را تضمین میکند.
حالت SE (Single-End) زمانی استفاده میشود که دادههای توالییابی شما تنها از یک سمت مولکول DNA خوانده شده باشند. در این حالت، شما یک فایل ورودی به Trimmomatic میدهید. حالت PE (Paired-End) زمانی به کار میرود که توالییابی از دو سمت مولکول DNA انجام شده باشد. در این صورت، شما باید دو فایل ورودی (یک فایل برای خوانشهای اول و یک فایل برای خوانشهای دوم) به Trimmomatic بدهید.

تیم تولید محتوای گروه بیوانفورماتیک وانیار در تلاش است تا بهترین آموزشهای کوتاه در زمینه بیوانفورماتیک و زیستشناسی را تهیه نماید. صحت محتوای این صفحه توسط کارشناسان گروه بیوانفورماتیک وانیار بررسی شده است.




عضویت در مجله وانیار
چطور از جدیدترین آموزشها باخبر شوم؟
با عضویت در مجله بیوانفورماتیک وانیار، برترین آموزشهای بیوانفورماتیک را در لحظه انتشار دریافت کنید.
سلام، وقت بخیر.
چطور میتونیم بهتون کمک کنیم؟
تیم ما آماده پاسخگویی به سوالات شماست.
پشتیبانی 24 ساعته در 7 روز هفته.


