
Jalview یک نرمافزار رایگان، متنباز (Open-source) و بسیار قدرتمند است که بهطور تخصصی برای ویرایش، نمایش و تحلیل توالیهای زیستی طراحی شده است. این نرمافزار در دنیای بیوانفورماتیک به عنوان یک ابزار استاندارد برای کار با همردیفی توالیها (Multiple Sequence Alignment – MSA) شناخته میشود.
نسخهٔ اصلی Jalview توسط Michele Clamp و همکارانش در سال 1996 توسعه داده شد، و Jalview 2 (نسخهٔ بازطراحیشده و بازنویسیشده) در سال 2005 منتشر شد.
در این مقاله، قصد داریم شما را با ویژگیهای گرافیکی و قابلیتهای کاربردی Jalview آشنا کنیم و نشان دهیم چگونه میتوانید از این ابزار برای پروژههای تحقیقاتی و آموزشی خود بهره ببرید.
نصب Jalview بسیار ساده است و روی تمام سیستمعاملهای اصلی (ویندوز، مک و لینوکس) قابل اجراست. برای شروع، نسخه سازگار با سیستمعامل خود را از لینکهای ارائهشده دانلود نمایید.
رمز فایل فشرده: www.vanyarbioinf.ir
پس از دانلود فایل نصبی، با اجرای آن و دنبال کردن دستورالعملهای ساده، نرمافزار روی سیستم شما نصب میشود. توجه داشته باشید که پیشنیاز اصلی Jalview، نصب Java Runtime Environment (JRE) است. در صورتی که این پیشنیاز بر روی سیستم شما نصب نباشد، میتوانید آن را بهطور جداگانه از وبسایت رسمی Java دریافت و نصب کنید.
Jalview میتواند فایلهای همترازی توالی را در قالبهای رایج مثل Fasta ، ClustalW (ALN) ، GCG-MSF ، PIR ، Pfam/Stockholm و چند فرمت استاندارد دیگر باز کند. برای باز کردن فایل میتوانید از کلید میانبر Ctrl+O یا منوی File→Input Alignment استفاده کنید و فایل را از سیستم خود، یک آدرس اینترنتی (با وارد کردن لینک کامل) یا از طریق کپیکردن متن در کادر مربوطه وارد کنید. برنامه معمولاً نوع فایل را بهطور خودکار تشخیص میدهد و اگر فرمت ناشناخته باشد یا خطایی وجود داشته باشد، پیام خطا نمایش میدهد. همچنین امکان بارگذاری پروژههای ذخیرهشده Jalview (شامل همترازیها، رنگبندی، توضیحات و درختها) از طریق File→Load Project وجود دارد.
یکی از قابلیتهای منحصربهفرد Jalview، امکان دسترسی مستقیم به دیتابیسهای بیولوژیکی آنلاین است.
بخش Sequence Fetcher در جالویو این امکان را میدهد که توالیها را مستقیماً از پایگاههای معتبر زیستی مانند UniProt ، PDB ، Ensembl ، EMBL ، PFAM و RFAM دریافت کنید. این ابزار را میتوان از مسیر File→Fetch Sequences در صفحه اصلی برای ساخت یک الاینمنت جدید، یا از داخل یک الاینمنتِ بازشده برای افزودن توالیهای بیشتر اجرا کرد. هنگام باز شدن اولیه ممکن است کمی مکث ایجاد شود، چون برنامه به وبسرویس دیتابیسها متصل میشود. هر بار که پنجره Fetcher باز میشود، باید دیتابیس موردنظر را از فهرست انتخاب کنید.
برای دریافت توالیها دو روش وجود دارد: در برخی پایگاهها مثل UniProt و PDB میتوانید با جستجوی متنی (بر اساس نام، توضیح یا شناسه) دیتا را پیدا کنید. در روش دیگر کافی است یک یا چند شناسه (accession ID) را وارد کنید تا توالیها بازیابی شوند. در پایان با زدن دکمه OK فرآیند دریافت انجام میشود.
این ویژگی باعث صرفهجویی قابلتوجهی در زمان میشود و نیاز به استفاده از چندین نرمافزار مختلف برای انجام وظایف مرتبط را برطرف میکند.
با استفاده از Jalview هم میتوانید الاینمنت را اجرا کنید و هم ظاهر نمایش توالیها را مطابق نیاز تغییر دهید:
Jalview این امکان را فراهم میکند تا بدون نیاز به خروج از نرمافزار، الاینمنتهای چندگانه توالی را انجام دهید. از طریق مسیر Web Service → Alignment، میتوانید به سرویسهای همترازسازی آنلاین مانند Clustal Omega ، MUSCLE و T-Coffee دسترسی داشته باشید. رابط گرافیکی Jalview به شما اجازه میدهد تا پارامترهای الگوریتم را بهراحتی تنظیم کنید و پس از اتمام فرآیند، نتایج همترازسازی بهصورت گرافیکی و با رنگآمیزی مناسب نمایش داده میشوند.
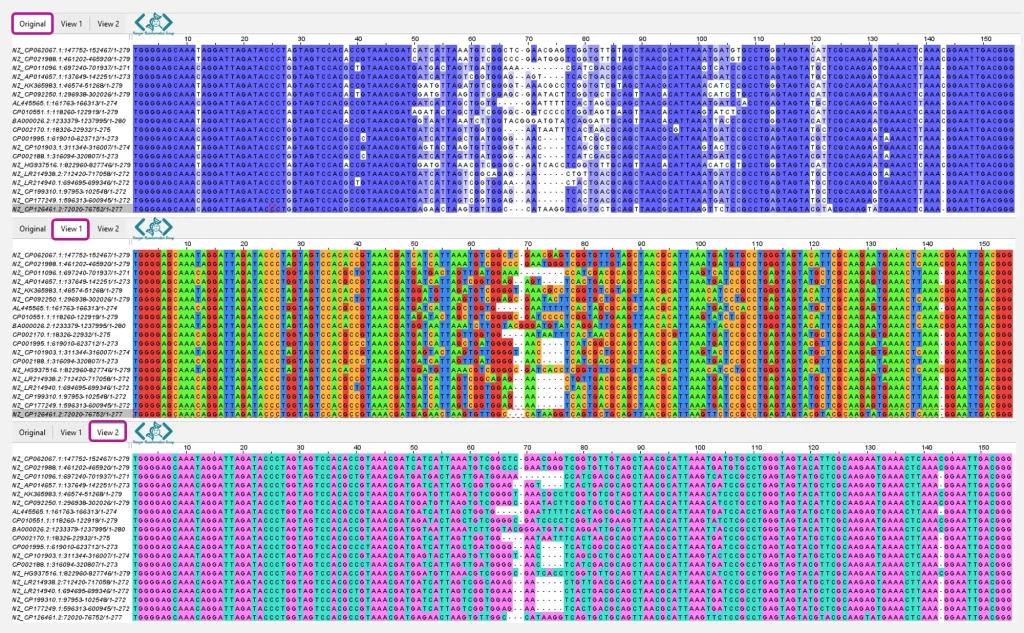
Jalview به شما امکان میدهد تا یک همترازی را به روشهای مختلف و همزمان مشاهده کنید. هر “نما” (view) یک نمایش مستقل از همان همترازی است که میتوانید تنظیمات مربوط به ترتیب نمایش توالیها، رنگبندی، پنهان کردن ردیفها یا ستونها، و نمایش ویژگیها و حاشیهنویسیها (annotations) را بهطور جداگانه برای آن تغییر دهید. با این حال، هرگونه ویرایش در خود دادههای همترازی (مانند تغییر ترتیب توالیها یا حاشیهنویسیها) بر روی تمام نماها اعمال خواهد شد.
برای ایجاد یک نمای جدید، میتوانید از منوی “View→New View” یا کلید میانبر Control+T استفاده کنید. نماهای جدید در ابتدا شبیه نمای اصلی هستند، اما تغییرات بعدی در ظاهر آنها، فقط روی همان نما تأثیر میگذارد. میتوانید با کلیک راست روی تب نما، آن را نامگذاری کرده و برای تمرکز بر بخش خاصی از همترازی، قسمتهای دیگر را مخفی کنید. بهطور پیشفرض، این نماها بهصورت تبهایی در یک پنجره واحد جمع میشوند، اما با استفاده از “View→Expand” (یا کلید X) میتوانید هر نما را در پنجرهای جداگانه باز کنید و با کلید G یا “View→Gather” دوباره آنها را در یک پنجره جمع کنید.
همچنین، درختهای محاسباتی، نتایج PCA و نمایش ساختارهای سهبعدی مرتبط با یک نما، میتوانند با نماهای دیگر نیز اشتراکگذاری شوند.
در ادامه، امکانات Jalview برای درک عمیقتر ساختار پروتئینها، شامل پیشبینی ساختار دوم و تجسم ساختارهای سهبعدی، مورد بررسی قرار میگیرد.
JPred در Jalview میتواند ساختار دوم پروتئینها را پیشبینی کند. ساختار دوم شامل الگوهای تکرارشوندهای مثل مارپیچ آلفا (alpha-helix) و صفحه بتا (beta-sheet) است که به شکل کلی پروتئین کمک میکنند.
JPred با استفاده از شبکههای عصبی و مقایسه توالی پروتئین شما با توالیهای دیگر که ساختار دومشان از قبل مشخص شده، حدس میزند که بخشهای مختلف پروتئین شما چه ساختاری خواهند داشت. این کار به درک بهتر عملکرد پروتئین کمک میکند.
🟥 از مسیر زیر به JPred دسترسی خواهید داشت:
Web Service→Secondary Structure Prediction→JPred Secondary Structure Prediction
نکته مهم این است که JPred ساختار دوم را بر اساس بخشهای پیوسته توالی پیشبینی میکند. اگر ستونهایی را مخفی کرده باشید، ممکن است دقت پیشبینی در نزدیکی آن مرزها کمی کمتر باشد. نتایج بهصورت یک پنجره جدید نمایش داده میشوند که ساختارهای پیشبینیشده را بههمراه اطلاعاتی درمورد میزان اطمینان پیشبینی نشان میدهد.
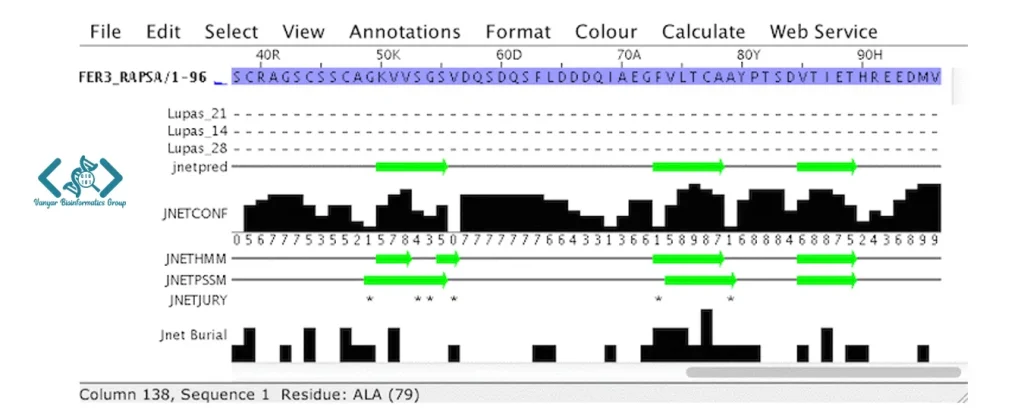
در زیرِ الاینمنت، نوارهایی از توضیحات (annotation) نمایش داده میشود که هر کدام معنی خاصی دارند:
* دیده شود، یعنی وقتی پیشبینیهای اولیه اختلاف زیادی داشتهاند، یک سیستم داوری برای انتخاب بهترین نتیجه به کار رفته است.Jalview ابزار جالبی برای مشاهده و تحلیل ساختارهای سهبعدی پروتئینها است 🥸
با استفاده از قابلیت 3D Structure Data، میتوانید مستقیماً ساختارهای پروتئینی را از دیتابیسهای معتبر مانند PDB و 3D-Beacons Network جستجو، انتخاب و دریافت کنید. Jalview بهطور خودکار بهترین ساختار را شناسایی کرده و یا به شما اجازه میدهد تا ساختارهای مورد نظر را بهصورت دستی انتخاب کرده یا حتی از فایلهای محلی (PDB/mmCIF) وارد نمایید.
پس از انتخاب ساختار، میتوانید آن را در نمایشگرهای مختلفی مانند Jmol ، Chimera ، ChimeraX و PyMOL مشاهده کنید. این نرمافزار قابلیتهای پیشرفتهای مانند تراز کردن (superimpose) ساختارها بر اساس توالیهای همتراز را نیز دارد تا بتوانید شباهتها و تفاوتهای ساختاری را بهدقت بررسی کنید.
علاوه بر این، Jalview امکان مشاهده و تفسیر شمارهگذاری آمینواسیدهای هر پروتئین در PDB و همچنین استفاده از دادههای ساختاری برای تزیین توالی با اطلاعاتی مانند ساختار دوم (secondary structure) و فاکتور دما (temperature factor) را فراهم میآورد.
Jalview این امکان را فراهم میکند که بر اساس همترازیهای چندگانه، درختهای فیلوژنتیک بسازید و روابط شباهت یا فاصله میان توالیها را بررسی کنید. به این منظور، از منوی Calculate گزینه Calculate Tree, PCA or PaSiMap را انتخاب کنید تا به پنجره محاسبات دسترسی پیدا کنید.
در این پنجره، شما میتوانید:
۱. نوع تحلیل مورد نظر خود را انتخاب کنید:
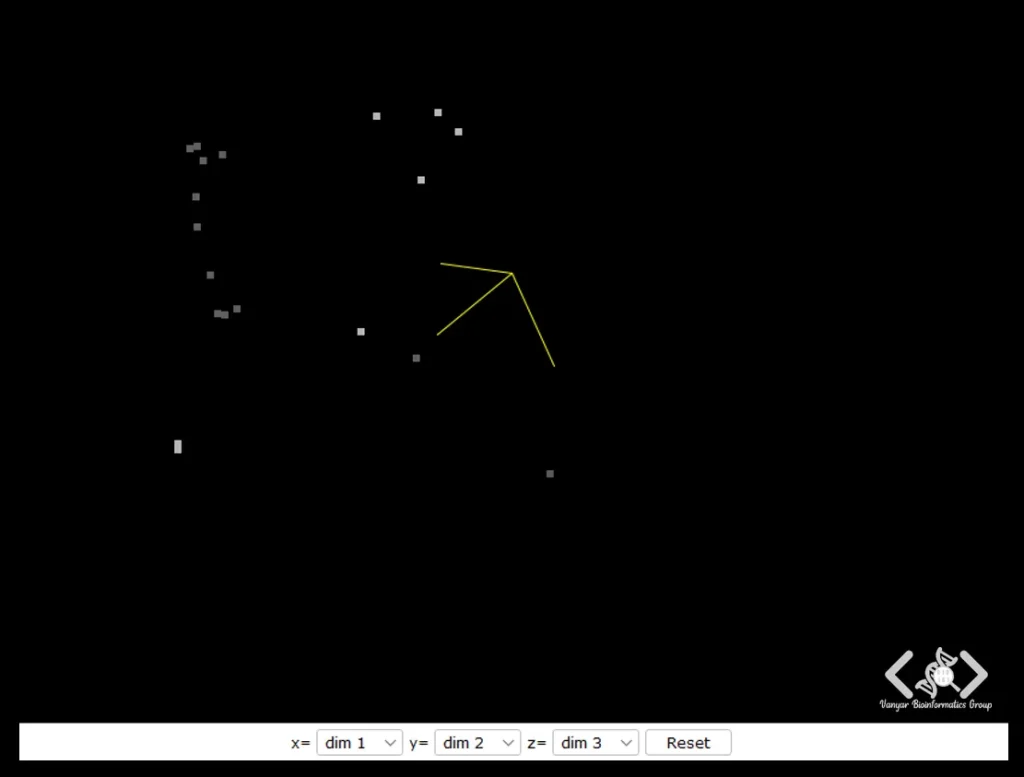
PCA یا Principal Component Analysis : روشی است برای نمایش شباهت بین توالیها در یک فضای سهبعدی؛ بهطوریکه توالیهای شبیه به هم نزدیکتر به هم قرار میگیرند.
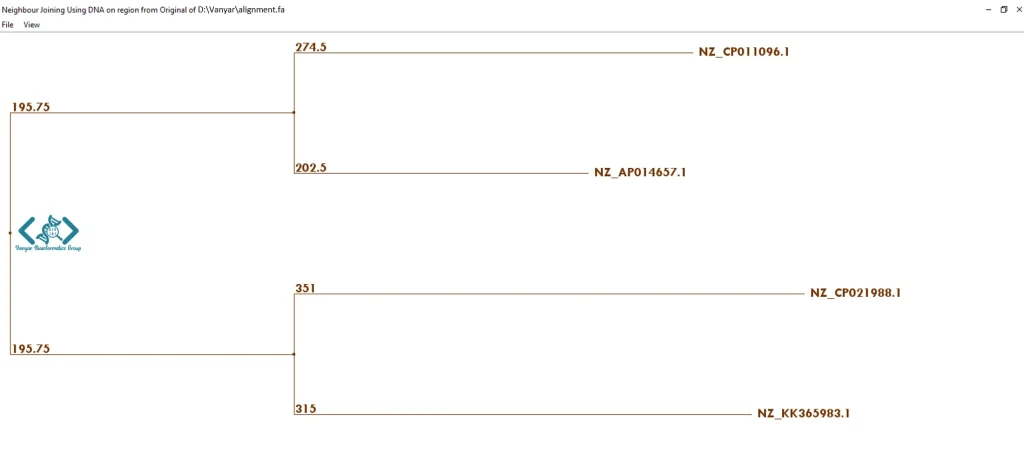
Tree : در Jalview میتوان درختهای شباهت/فاصله را از روی همترازی توالیها ساخت؛ این کار یا روی کل alignment انجام میشود یا فقط روی توالیهای انتخابشده. برای ساخت درخت فیلوژنتیکی دو روش اصلی وجود دارد: UPGMA که سادهتر و سریعتر است، و Neighbour Joining که دقیقتر ولی از نظر محاسباتی سنگینتر است.
PaSiMap : مانند PCA، توالیها را در یک فضای سهبعدی نمایش میدهد تا شباهتها را نشان دهد. تفاوت اصلی آن این است که به جای استفاده از امتیازات شباهت از یک همترازی چندتایی، PaSiMap ابتدا یک الاینمنت دوتایی برای هر جفت توالی محاسبه میکند، که این روش برای توالیهای بسیار نزدیک مؤثرتر است.
همترازی دوتایی (Pairwise Alignment) : این روش برای مقایسه دقیق شباهت بین هر جفت توالی استفاده میشود و نتایج آن در قالب یک پنجره متنی نمایش داده میشود.

۲. مدل امتیاز شباهت توالی را انتخاب کنید: در قسمت پایین پنجره، گزینههایی برای انتخاب نحوه محاسبه شباهت بین توالیها وجود دارد که برای انجام تحلیل انتخابی شما استفاده خواهد شد. میتوان از معیارهایی مثل PID، ماتریسهای جایگزینی مثل BLOSUM62 / PAM250 / DNA یا شباهت بر اساس ویژگیهای توالی و حتی ساختار ثانویه استفاده کرد.
Jalview گزینههای متنوعی برای ذخیره و خروجی گرفتن از دیتا و نتایج تحلیلها ارائه میدهد.
برای ذخیره الاینمنتها، میتوانید از مسیر File→Save As آنها را در قالبهای استاندارد مختلف مانند PNG ، SVG و EPS ذخیره کنید یا اجازه دهید برنامه فرمت مناسب را بر اساس پسوند فایل انتخاب کند. تنظیمات پیشفرض Jalview شامل افزودن شماره شروع و پایان توالی به نام آن است که قابل تغییر میباشد. همچنین امکان خروجی گرفتن از توضیحات و نمادهای همترازی بهصورت فایل CSV وجود دارد. برای حفظ کامل جزئیات کار، از جمله همترازیها، رنگبندیها، انوتیشنها، درختها و وضعیت نمایش ساختارهای سهبعدی، میتوانید پروژه را بهصورت یک فایل آرشیوی .jvp از طریق File→Save Project as ذخیره کرده و در آینده بازیابی کنید. این قابلیتها برای ادامه کار، اشتراکگذاری نتایج و ارائه به دانشجویان بسیار کاربردی هستند.
در مقایسه با سایر نرمافزارهای تحلیل توالی، Jalview مزایای قابل توجهی دارد:
🔴 اول، رابط کاربری گرافیکی غنی و در عین حال کاربرپسند آن است که کار با دادههای پیچیده توالی را آسان میکند. Jalview نیازی به دانش برنامهنویسی ندارد و برای دانشجویان و محققانی که تمرکز اصلی آنها بر بیولوژی است، مناسبتر است.
🟣 دوم، یکپارچگی گسترده Jalview با سرویسهای وب و دیتابیسهای آنلاین است که دسترسی به ابزارها و دادههای متنوع را بدون خروج از محیط نرمافزار فراهم میکند.
🟠 سوم، قابلیتهای تجسمسازی پیشرفته آن است که امکان نمایش همزمان دیتای توالی، ساختاری و تکاملی را با جزئیات کامل فراهم میکند.
Jalview با ترکیب رابط کاربری گرافیکی قدرتمند، قابلیتهای تحلیلی متنوع و محیطی کاربرپسند، به ابزاری کاربردی برای مشاهده، ویرایش و تحلیل همترازیهای چندگانه توالی تبدیل شده است. این نرمافزار با پشتیبانی از یکپارچهسازی با سرویسهای وب، نمایش همزمان توالی و ساختار، و ابزارهای پیشرفته تجسم دیتا، فراتر از یک ویرایشگر ساده عمل میکند. رایگان و متنباز بودن Jalview نیز آن را به گزینهای پایدار و قابل اعتماد برای پژوهش، آموزش و تحلیلهای بیوانفورماتیکی مختلف تبدیل کرده است.
Jalview یک نرمافزار رایگان و متنباز برای مشاهده، ویرایش و تحلیل همترازی توالیهای زیستی مانند DNA ، RNA و پروتئین است. این برنامه در بیوانفورماتیک برای بررسی توالیها و تحلیلهای تکاملی بسیار پرکاربرد است.
بله، Jalview برای اجرا به Java Runtime Environment یا JRE نیاز دارد. اگر Java روی سیستم نصب نباشد، باید ابتدا آن را نصب کنید.
Jalview میتواند به دیتابیسهای آنلاین مانند UniProt ، PDB ، Ensembl ، EMBL ، PFAM و RFAM متصل شود. این قابلیت، دریافت سریع توالیها را بدون نیاز به خروج از برنامه ممکن میکند.
بله، Jalview امکان اجرای الاینمنت چندتایی (MSA) را از طریق سرویسهای آنلاین مانند Clustal Omega ، MUSCLE و T-Coffee فراهم میکند. نتیجه همترازی نیز بهصورت گرافیکی و قابل تنظیم نمایش داده میشود.
JPred ساختار دوم پروتئین را بر اساس توالی آن پیشبینی میکند و بخشهایی مانند مارپیچ آلفا و صفحه بتا را نشان میدهد. نتیجه پیشبینی همراه با میزان اطمینان نمایش داده میشود.

تیم تولید محتوای گروه بیوانفورماتیک وانیار در تلاش است تا بهترین آموزشهای کوتاه در زمینه بیوانفورماتیک و زیستشناسی را تهیه نماید. صحت محتوای این صفحه توسط کارشناسان گروه بیوانفورماتیک وانیار بررسی شده است.




عضویت در مجله وانیار
چطور از جدیدترین آموزشها باخبر شوم؟
با عضویت در مجله بیوانفورماتیک وانیار، برترین آموزشهای بیوانفورماتیک را در لحظه انتشار دریافت کنید.
سلام، وقت بخیر.
چطور میتونیم بهتون کمک کنیم؟
تیم ما آماده پاسخگویی به سوالات شماست.
پشتیبانی 24 ساعته در 7 روز هفته.



