با ظهور تکنولوژیهای توالییابی نسل جدید (NGS)، حجم دادههای تولیدشده در آزمایشگاههای ژنتیک به شکل چشمگیری افزایش یافت. در این میان، Samtools به عنوان یکی از پرکاربردترین مجموعه ابزارهای بیوانفورماتیک برای مدیریت، پردازش و تحلیل این دادههای حجیم پا به عرصه گذاشت.
این نرمافزار متنباز که در حال حاضر بهعنوان یک استاندارد جهانی در آزمایشگاههای بیوانفورماتیک شناخته میشود، برای تعامل با دادههای همترازی توالیها (Sequence Alignments) طراحی شده است. توسعهدهندگان این ابزار همواره تلاش کردهاند تا با استانداردهای گروه کاری GA4GH (اتحاد جهانی برای ژنومیک و سلامت) همگام باشند و فرمتهای استانداردی را برای ذخیره و پردازش دادهها ارائه دهند. با درک عمیق از Samtools، محققان حوزه ژنومیک میتوانند پایپلاینهای تحلیلی خود را بهینهتر کرده و به نتایج دقیقتری دست یابند.
ریشههای تاریخی Samtools به سال ۲۰۰۹ و پروژه ۱۰۰۰ ژنوم بازمیگردد. در آن زمان، Heng Li و همکارانش برای اولین بار فرمت SAM (Sequence Alignment/Map) و ابزار خط فرمان Samtools را در ژورنال Bioinformatics معرفی کردند. هدف اصلی این بود که راه حلی کارآمد برای مدیریت حجم عظیمی از دادههای مپشده (Mapped data) ایجاد شود.
با گذشت زمان و گسترش نیازهای تحلیلی، این پروژه از یک ابزار ساده به یک اکوسیستم سهگانه و قدرتمند تبدیل شد. در سال ۲۰۲۱، مقالهای جامع درباره دستاوردها و پیشرفتهای ۱۲ ساله این مجموعه نرمافزاری در ژورنال GigaScience منتشر شد که نشاندهنده پویایی و توسعه مداوم آن است. امروزه وقتی از Samtools صحبت میکنیم، در واقع با سه بخش مجزا اما کاملاً یکپارچه روبرو هستیم:
این تفکیک هوشمندانه باعث شده است تا هر ابزار با بالاترین سرعت و کمترین میزان مصرف حافظه، وظیفه تخصصی خود را انجام دهد. این مجموعه نرمافزاری تا به امروز نقش حیاتی در موفقیت پروژههای بزرگ ژنومی مانند ENCODE و پروژه اطلس ژنوم سرطان (TCGA) ایفا کرده است.
نصب نرمافزار Samtools در سیستمعاملهای مختلف نسبتاً ساده است و از طریق مدیر بستههای (Package Managers) استاندارد به راحتی انجام میشود. با این حال، اگر با حجم عظیمی از دادههای ژنومی سر و کار دارید، نحوه نصب شما میتواند تأثیر چشمگیری روی سرعت پردازش نهایی داشته باشد.
امروزه استانداردترین روش برای نصب نرمافزارهای بیوانفورماتیک، استفاده از Bioconda است. مزیت بزرگ این روش این است که تمام پیشنیازها (Dependencies) را بهطور خودکار نصب میکند و نیازی به دسترسی ادمین (sudo) ندارد. اگر آناکوندا یا مینیکوندا روی سیستم شما نصب است، با اجرای دستور زیر، Samtools به راحتی نصب میشود:
conda install -c bioconda samtools
(نکته: برای سرعت بیشتر در نصب پکیجها، استفاده از mamba به جای conda نیز به شدت پیشنهاد میشود).
اگر فقط قصد دارید Samtools را برای کارهای روزمره و فایلهای نهچندان حجیم نصب کنید، دستورات زیر کار شما را راه میاندازند:
در توزیعهای مبتنی بر Ubuntu/Debian:
sudo apt-get install samtools در سیستمعامل Mac OS (با استفاده از Homebrew):
brew install samtools کاربران ویندوز نیز میتوانند از طریق محیط WSL (Windows Subsystem for Linux) این ابزار را نصب کرده و از خط فرمان لینوکس در ویندوز بهرهمند شوند.
از آنجا که دادههای توالییابی بسیار فشرده هستند، نرمافزار Samtools بخش زیادی از زمان پردازش را صرفِ فشردهسازی و بازگشایی این اطلاعات (Encoding/Decoding) میکند.
در حالت عادی، Samtools از کتابخانه استاندارد Zlib استفاده میکند. اما توسعهدهندگان در مستندات رسمی HTSlib اکیداً توصیه میکنند که از کتابخانه libdeflate استفاده کنید. بنچمارکهای رسمی ثابت میکنند که کتابخانه libdeflate سرعت خواندن و نوشتن فایلها را به شکل چشمگیری افزایش میدهد.
چگونه Samtools را برای حداکثر سرعت کامپایل کنیم؟
برای راحتی کار، ما سورسکد کامل Samtools را برای دانلود مستقیم قرار دادهایم. شما باید ابتدا فایل زیر را دانلود کنید و بعد آن را از حالت فشرده نمایید.
رمز فایل فشرده: www.vanyarbioinf.ir
پیش از شروع کامپایل، باید مطمئن شوید که کتابخانه libdeflate (و بستههای توسعه آن مانند libdeflate-dev) روی سرور یا سیستم شما نصب شده باشد. وقتی این پیشنیاز را نصب کردید، وارد پوشه سورسکد شوید و نرمافزار Samtools را با دستورات زیر برای حداکثر سرعت پیکربندی و نصب کنید.
cd samtools-source-directory
./configure --with-libdeflate
make
sudo make installپس از پایان نصب، میتوانید با اجرای دستور samtools --version از نصب صحیح برنامه و ماژولهای فعال آن اطمینان حاصل کنید. با این روش نصب، شما یک موتور پردازشی قدرتمندتر خواهید داشت که در پروژههای بزرگ (مثل WGS) زمان اجرای پایپلاینهای شما را ساعتها کاهش میدهد.
دستورات در Samtools از یک ساختار منطقی و یکپارچه پیروی میکنند که یادگیری آنها را بسیار آسان میکند. الگوی کلی دستورات به این شکل است:
samtools [command] [options] [input_file] [output_file]اما در پروژههای واقعی بیوانفورماتیک، ما معمولاً دستورات را به صورت تکی اجرا نمیکنیم. مستندات رسمی Samtools اکیداً توصیه میکند که به جای تولید فایلهای موقت در هر مرحله، از پایپلاینهای یونیکس (استفاده از علامت |) استفاده کنید. این کار باعث میشود خروجی هر مرحله مستقیماً به عنوان ورودی مرحله بعد خوانده شود و سرعت پردازش به شکل چشمگیری افزایش یابد. در تمام دستورات Samtools، علامت - به عنوان جایگزینی برای ورودی استاندارد (stdin) و خروجی استاندارد (stdout) شناخته میشود.
در ادامه، مهمترین دستورات را در قالب یک ورکفلوی استاندارد از فایل خام تا فایل نهایی بررسی میکنیم:
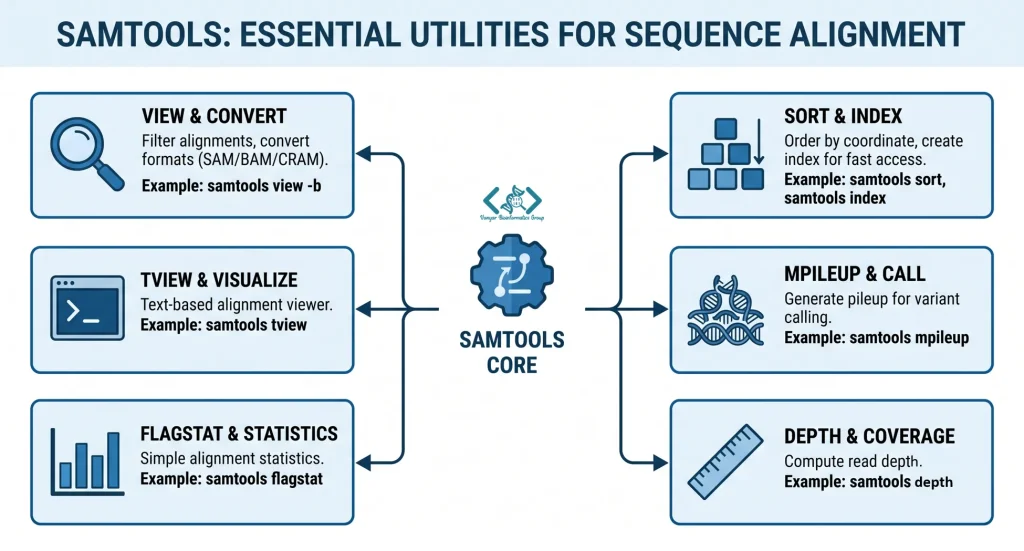
دستور view آچار فرانسه Samtools است. از این دستور برای تبدیل فرمتها (مثلاً SAM به BAM یا CRAM) و همچنین فیلتر کردن خوانشها استفاده میشود. برای افزایش سرعت در پایپلاینها، توسعهدهندگان پیشنهاد میکنند از گزینه -u (یا -O bam,level=0) استفاده کنید تا خروجی به صورت BAM غیرفشرده تولید شود. چون این دادهها روی دیسک ذخیره نمیشوند و مستقیماً به مرحله بعد میروند، حذف فرآیند فشردهسازی باعث صرفهجویی زمان میشود.
یکی از مراحل بسیار مهمی که در پایپلاینهای حرفهای انجام میشود، استفاده از دستور fixmate است. این ابزار وظیفه دارد هرگونه نقص یا خطایی که نرمافزار همترازکننده (Aligner) در جفتخوانشها (Mate-pairs) ایجاد کرده است را اصلاح کند. در واقع این مرحله به عنوان یک گام «غلطگیری» (Proof-reading) عمل میکند تا مطمئن شویم اطلاعات مربوط به جهتگیری خوانشها و فاصلهها کاملاً با یکدیگر همخوانی دارند.
بسیاری از ابزارهای پاییندستی نیازمند فایلهایی هستند که بر اساس موقعیت ژنومی مرتب شده باشند. دستور sort این کار را انجام میدهد. از آنجا که مرتبسازی یک فرآیند به شدت موازی است، میتوانید با استفاده از پارامتر -@ تعداد رشتههای پردازشی (Threads) را مشخص کنید تا کار با سرعت بیشتری انجام شود. همچنین دقت کنید که اگر با استفاده از پارامتر -m حافظه بیشتری به این دستور اختصاص میدهید، این مقدار حافظه به ازای هر رشته پردازشی محاسبه میشود، نه کل فرآیند.
پس از ایجاد فایل مرتبشده نهایی، ایجاد فایل ایندکس (با دستور index) کاملاً ضروری است. این فایل که با پسوند .bai یا .crai تولید میشود، مانند فهرست یک کتاب عمل میکند و به نرمافزارهای مصورسازی مانند IGV اجازه میدهد بدون نیاز به خواندن کل فایل، مستقیماً به یک ژن یا ناحیه خاص پرش کنند. همچنین اگر دادههای یک نمونه در چندین لاین (Lane) مختلف توالییابی شده باشند، میتوانید آنها را با استفاده از دستور merge در یک فایل واحد ادغام کنید.
در اینجا نمونهای از یک اسکریپت بهینه را میبینید که فایل خام را همتراز کرده (در اینجا با minimap2)، جفتخوانشها را اصلاح نموده، فایل را مرتب کرده و در نهایت به فرمت فوقفشرده CRAM تبدیل میکند، بدون اینکه هیچ فایل موقتی روی هارد دیسک شما ساخته شود:
minimap2 -a reference.fa read1.fq read2.fq | \
samtools fixmate -u -m - - | \
samtools sort -u -@ 8 - | \
samtools view -C -T reference.fa -@ 8 -o final_output.cram -
با اجرای این پایپلاین یکپارچه، شما نهتنها از درگیر شدن بیمورد هارد دیسک جلوگیری کردهاید، بلکه زمان اجرای تحلیلهای بیوانفورماتیکی خود را که در پروژههای بزرگ ممکن است ساعتها طول بکشد، به حداقل رساندهاید.
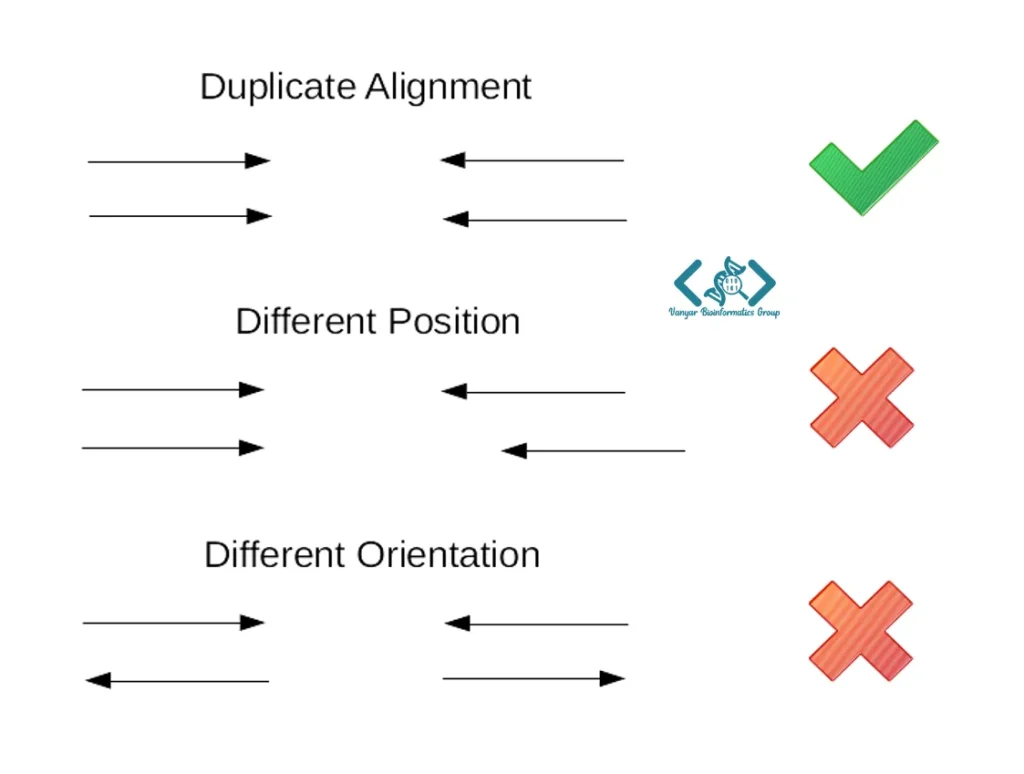
در فرآیند توالییابی، گاهی دستگاه توالییاب یک مولکول اولیه را بیش از حد میخواند که متخصصان به این حالت «خوانش تکراری» میگویند. وجود این خوانشها باعث میشود حضور یک توالی خاص به اشتباه در دادهها بیشنمایی (Over-representation) شود و نتایج تحلیل را دچار سوگیری کند. به طور کلی، دو نوع اصلی از این دادههای تکراری وجود دارد:
منطق Samtools برای پیدا کردن تکرارها بسیار هوشمندانه است. از آنجا که این خوانشها کپیهای دقیقی از هم هستند، پس از همترازی دقیقاً در یک موقعیت مختصاتی روی ژنوم مرجع و با یک جهتگیری یکسان قرار میگیرند. همانطور که در تصویر زیر میبینید، اگر دو یا چند خوانش مقادیر کاملاً یکسانی در موقعیت و جهتگیری داشته باشند، خوانشی که بالاترین کیفیت بازها را داشته باشد به عنوان «نسخه اصلی» حفظ شده و بقیه به عنوان «تکراری» (Duplicate) در نظر گرفته شده و با فعال شدن فلگ DUP در فایل همترازی علامتگذاری میشوند.
*** اگر برای شما مهم است که بدانید کدام تکرارها از نوع Optical بودهاند، Samtools پارامتری به نام -d به معنای Distance را ارائه کرده است. این پارامتر فاصله فیزیکی کلاسترها روی Flow Cell را بررسی میکند.
بسیاری از کاربران در ابتدا تصور میکنند فقط با اجرای دستور samtools markdup کار تمام است! اما بر اساس مستندات رسمی، این ابزار برای کار کردن به اطلاعات دقیقی از خوانشهای جفتشده (Mate-pairs) نیاز دارد. بنابراین، ورکفلوی استاندارد باید شامل 4 مرحله باشد:
collate): خوانشها را بر اساس نامشان کنار هم قرار میدهد. fixmate -m): این مرحله حیاتی است! پارامتر -m تگهای MC (رشته سیگارِ جفت) و ms (امتیاز کیفیت جفت) را به فایل اضافه میکند که ابزار markdup در مرحله آخر برای محاسباتش به آنها نیاز قطعی دارد. sort): فایل را بر اساس موقعیت روی ژنوم مرتب میکند. markdup): در نهایت تکرارها شناسایی میشوند. برای جلوگیری از ساخت فایلهای موقت و افزایش حداکثری سرعت، میتوانید این 4 مرحله را با استفاده از پایپلاین یونیکس و پارامتر -u (برای جلوگیری از فشردهسازی در مراحل میانی) به شکل زیر ترکیب کنید:
samtools collate -O -u example.bam | \
samtools fixmate -m -u - - | \
samtools sort -u - | \
samtools markdup -f stats_file.txt -S -d 2500 - final_markdup.bam
با اجرای این دستور، یک فایل خروجی تمیز دریافت میکنید و یک فایل آماری (stats_file.txt) نیز ساخته میشود که جزئیات دقیقی از تکرارهای یافت شده را به شما گزارش میدهد.
فرآیند استخراج واریانتهای ژنتیکی (SNPها و InDelها) معمولاً یک فرآیند دو مرحلهای است. در پایپلاینهای استاندارد، Samtools با استفاده از دستور mpileup وظیفه جمعآوری، تجمیع و بررسی اطلاعات الاینمنت در هر موقعیت ژنومی را بر عهده دارد. پس از اینکه Samtools این دادههای خام را آماده کرد، خروجی آن را برای فراخوانی نهایی به نرمافزار دیگری (معمولاً BCFtools) میدهیم.
شکل استاندارد این همکاری به صورت زیر است:
samtools mpileup -uf reference.fasta sample.bam | bcftools call -mv -o variants.vcf
مفهوم «فیلترینگ پیش از فراخوانی»، یکی از مهمترین مفاهیمی است که توسعهدهندگانِ Samtools در مستندات رسمی خود آن را بررسی کردهاند. در این روش، برنامه قبل از اینکه اصلاً خطی را در فایل VCF نهایی تولید کند، تصمیم میگیرد که آیا یک واریانت ارزش کاندید شدن دارد یا خیر.
این نوع فیلترینگ از طریق پارامترهای دستور samtools mpileup انجام میشود و نقش بسیار مهمی در کاهش خطاهای دستگاه توالییاب (Artifacts) دارد.
شما میتوانید با اضافه کردن فلگهای زیر به دستور mpileup، دقت کار خود را به شدت بالا ببرید:
-m : این گزینه حداقل تعداد خوانشهای دارای فاصله (Gapped reads) را برای کاندید کردن یک InDel مشخص میکند. مقدار پیشفرض آن در نسخههای جدید ۲ است. -L : حداکثر عمق پوشش (Depth) مجاز برای خروجی دادن یک InDel را تعیین میکند. (دادههایی با عمق غیرطبیعی و بسیار بالا معمولاً نشاندهنده خطای همترازی هستند).--indel-bias : یک پارامتر کلیدی برای کنترل حساسیت سیستم است؛ اعداد بالاتر باعث یافتن InDelهای بیشتر (با دقت کمتر) و اعداد پایینتر باعث یافتن InDelهای کمتر (اما بسیار دقیقتر) میشود. -h : نواحی هموپلیمر (تکرار یک نوکلئوتید خاص) همیشه برای دستگاههای توالییاب دردسرساز هستند. این ضریب مشخص میکند که چقدر احتمال دارد یک واریانت در ناحیه هموپلیمر، صرفاً یک خطای توالییابی باشد. مقدار پیشفرض آن ۵۰۰ است و اعداد کمتر نشاندهنده احتمال خطای بیشتر است.-I: اگر در پروژه خود اصلاً نیازی به بررسی InDelها ندارید و فقط دنبال SNPها هستید، این گزینه کل فرآیند جستجوی InDel را متوقف کرده و سرعت کار را بالا میبرد. تنظیم درست این پارامترها در Samtools باعث میشود که فایل ورودی به BCFtools بسیار تمیزتر و قابلاعتمادتر باشد.
تحلیل پوشش ژنومی برای ارزیابی کیفیت دادههای توالییابی و اطمینان از پوشش کافی در مناطق مورد علاقه بسیار مهم است.
*** تفاوت Depth و Coverage: اگرچه در محاورات بیوانفورماتیک این دو واژه غالباً به جای هم استفاده میشوند، اما از نظر تکنیکی متفاوتاند. عمق (Depth) به تعداد دفعاتی اشاره دارد که یک موقعیت خاص روی ژنوم توسط دستگاه خوانده شده است (مثلاً 50X)، در حالی که پوشش (Coverage) به درصدی از کل طول ژنوم یا ناحیه هدف اشاره دارد که توالییابی شده است (مثلاً پوشش ۹۹٪). دستور
samtools depthدر واقع عمق تکتک نقاط را محاسبه میکند تا به کمک آن بتوانیم تحلیل جامع پوشش ژنومی را انجام دهیم.
نواحی با پوشش بسیار بالا یا بسیار پایین میتوانند نشاندهنده مشکلات تکنیکی یا خصوصیات بیولوژیکی جالب باشند. یک افزایش ناگهانی و غیرطبیعی در عمق پوشش، معمولاً نشاندهنده خطای همترازی (Misalignment) یا وجود کپیهای تکراری در ناحیهای از ژنوم است. بنابراین، شناسایی این نواحی غیرعادی یک گام حیاتی در کنترل کیفیت دادهها محسوب میشود.
با استفاده از دستور depth در Samtools، میتوان عمق پوشش را استخراج کرد:
#محاسبه پوشش برای کل ژنوم:
samtools depth -a sample.bam > genome_coverage.txt
#تعیین کاوریج فقط برای یک منطقه خاص (مثلاً روی کروموزوم 1):
samtools depth -a -r chr1:1000000-2000000 sample.bam > region_coverage.txt#محاسبه و مقایسه پوشش همزمان برای چندین نمونه:
samtools depth -a sample1.bam sample2.bam sample3.bam > comparison_coverage.txtاستفاده از پارامتر -a در این دستورات بسیار مهم است، زیرا باعث میشود که موقعیتهای با پوشش صفر (نواحی که هیچ خوانشی ندارند) نیز در فایل خروجی گزارش شوند.
استخراج آمارهای پیشرفته با ترکیب Samtools و awk یکی از جذابیتهای کار است. شما میتوانید خروجی دستوراتی مثل depth را مستقیماً با ابزارهای پردازش متن مانند awk ترکیب کنید. این ترفند به شما اجازه میدهد آمارهای توصیفی قدرتمندی را استخراج کنید:
#محاسبه میانگین کل پوشش ژنوم:
samtools depth sample.bam | awk '{sum+=$3} END {print "Average coverage: " sum/NR}'#محاسبه درصدِ ژنوم که حداقل 10X پوشش دارد:
samtools depth -a sample.bam | awk -v X=10 '{if($3>=X) count++} END {print "Percentage covered by >=10X: " count/NR*100"%"}'#استخراج نواحی مشکوک با پوشش بسیار بالا (مثلاً بیشتر از 100X):
samtools depth sample.bam | awk '$3>100' > high_coverage_regions.txt
#استخراج نواحی با پوشش بسیار پایین (کمتر از 5X):
samtools depth -a sample.bam | awk '$3<5' > low_coverage_regions.txtبرای تحلیل پیشرفتهتر، میتوان از ابزارهای تخصصی مانند BEDTools استفاده کرد. به عنوان مثال، با استفاده از BEDTools میتوانید نواحی استخراجشده با پوشش پایین را با موقعیت ژنها همپوشانی دهید تا دقیقاً متوجه شوید کدام مناطق مهم پوشش مناسبی ندارند.
در حالی که Samtools سالهاست به عنوان استاندارد طلایی در پردازش فایلهای SAM/BAM شناخته میشود، اکوسیستم بیوانفورماتیک به سرعت در حال رشد است. با افزایش حجم دادهها در پروژههای WGS و ظهور توالییابیهای Long-read، ابزارهای جدیدی برای رفع گلوگاههای سرعتی و دقتی وارد میدان شدهاند. برای طراحی یک پایپلاین بهینه، شما باید رقبای Samtools را در سه دسته اصلی بشناسید:
اگر به دنبال ابزارهایی هستید که دقیقاً همان عملکردهای پایه Samtools را انجام دهند، این دو گزینه مطرح هستند:
این دسته از ابزارها بیشتر نقش «مکملهای قدرتمند» را برای Samtools بازی میکنند و در مطالعات بالینی به شدت محبوباند:
اکوسیستم مدرن بیوانفورماتیک در حال حرکت به سمت زبانهای برنامهنویسی جدید است تا محدودیتهای مدیریت حافظه در زبان C (زبان اصلی نرمافزار Samtools) را دور بزند:
Noodles یک بازنویسی کامل از HTSlib به زبان Rust است که بدون باگهای حافظه، پردازش فایلهای SAM/BAM/CRAM را با کارایی بالا انجام میدهد. همچنین ابزار Rustybam به طور ویژه برای دستکاری سریع دادههای توالییابی طولانی (مانند PacBio و Oxford Nanopore) طراحی شده است.samtools depth) باشد، Mosdepth رقیب بیرحم آن است. این ابزار که به زبان Nim نوشته شده، با استفاده از الگوریتمهای بهینه، فایلهای BAM/CRAM را دهها بار سریعتر از Samtools پردازش کرده و آمارهای پوشش را تولید میکند.نرمافزار Samtools بهعنوان یکی از پایههای اصلی تحلیل دادههای توالییابی، نقش بسیار مهمی در پیشرفتهای ژنومیک و تحقیقات پزشکی دقیق ایفا کرده است. این ابزار قدرتمند با ارائه مجموعهای از قابلیتهای بینظیر برای مدیریت، پردازش و استخراج اطلاعات از دادههای حجیم توالییابی، به محققان کمک میکند تا به بینشهای عمیقتری در مورد ژنوم انسان و سایر موجودات دست یابند.
در این مقاله، تلاش کردیم بررسی جامعی از Samtools، از دستورات پایهای تا ساخت پایپلاینهای پیشرفته و مقایسه آن با سایر غولهای بیوانفورماتیک داشته باشیم. تسلط بر این ابزار به شما اجازه میدهد تا ورکفلوهای پردازشی خود را به شکل چشمگیری بهینهسازی کرده و در نهایت به نتایجی دقیقتر و قابلاعتمادتر در پژوهشهای خود برسید. با پیشرفت سریع تکنولوژیهای توالییابی و تولید روزافزون دادههای عظیم (Big Data) در زیستشناسی، ابزارهای کارآمدی مانند اکوسیستم Samtools اهمیت به مراتب بیشتری پیدا میکنند و همچنان در مسیر تکامل و بهبود گام برخواهند داشت.
Samtools یک مجموعه ابزار قدرتمندِ خط فرمان در بیوانفورماتیک است که برای مدیریت، ویرایش، فیلتر کردن و استخراج اطلاعات آماری از فایلهای همترازی ژنومی (مانند فرمتهای SAM ، BAM و CRAM) استفاده میشود.
برای این کار باید از دستور view استفاده کنید. اجرای قطعه کد samtools view -b -o output.bam input.sam فایل متنی شما را به نسخه باینری و فشرده تبدیل میکند تا فضای کمتری در هارد سرور اشغال کند.
بله، معمولاً در پایپلاینهای بیوانفورماتیک، ابتدا با دستور samtools mpileup اطلاعات توالییابی در هر موقعیت ژنوم جمعآوری شده و سپس خروجی آن به نرمافزار BCFtools داده میشود تا واریانتها (SNPها و InDelها) استخراج شوند.
بله، Samtools یک نرمافزار کاملاً متنباز (Open-Source) و رایگان است. شما میتوانید بدون پرداخت هیچ هزینهای یا نگرانی بابت لایسنس، از آن در تمامی پروژههای دانشگاهی، تحقیقاتی و حتی محیطهای تجاری و بالینی استفاده کنید.
این ابزار روی سیستمعاملهای مبتنی بر یونیکس (مانند توزیعهای مختلف لینوکس و سیستمعامل Mac) اجرا میشود. کاربران ویندوز نیز برای استفاده از آن نیازی به تغییر سیستمعامل ندارند و میتوانند از طریق محیط WSL آن را به راحتی اجرا کنند.

تیم تولید محتوای گروه بیوانفورماتیک وانیار در تلاش است تا بهترین آموزشهای کوتاه در زمینه بیوانفورماتیک و زیستشناسی را تهیه نماید. صحت محتوای این صفحه توسط کارشناسان گروه بیوانفورماتیک وانیار بررسی شده است.




عضویت در مجله وانیار
چطور از جدیدترین آموزشها باخبر شوم؟
با عضویت در مجله بیوانفورماتیک وانیار، برترین آموزشهای بیوانفورماتیک را در لحظه انتشار دریافت کنید.
سلام، وقت بخیر.
چطور میتونیم بهتون کمک کنیم؟
تیم ما آماده پاسخگویی به سوالات شماست.
پشتیبانی 24 ساعته در 7 روز هفته.