نرمافزار Bowtie 2 یک ابزار فوقسریع و بهینه از نظر مصرف رم است که برای همترازی (Alignment) خوانشهای توالییابی (reads) با ژنومهای رفرنس طویل طراحی شده است. این ابزار به طور ویژه برای همترازی خوانشهایی با طول ۵۰ تا چند صد کاراکتر (جفتباز) بر روی ژنومهای نسبتاً بزرگ، مانند ژنوم پستانداران، عملکردی فوقالعاده دارد.
یکی از چالشهای همترازی، مصرف بالای منابع سیستم است؛ اما Bowtie 2 با ایندکسگذاری ژنوم بر اساس FM Index (مبتنی بر تبدیل Burrows-Wheeler یا BWT)، توانسته میزان مصرف رم را به حداقل برساند. به عنوان مثال، برای ژنوم انسان، میزان فضای اشغالشده در رم معمولاً تنها در حدود ۳.۲ گیگابایت است. علاوه بر این، پشتیبانی از پردازش چندگانه (Multi-threading) به شما اجازه میدهد تا با استفاده همزمان از چندین پردازنده، سرعت الاینمنت را به شکل چشمگیری افزایش دهید.
این ابزار از حالتهای مختلف همترازی از جمله موارد زیر پشتیبانی میکند:
خروجی استاندارد Bowtie 2 به فرمت SAM است که تعامل یکپارچه و آسان آن را با طیف وسیعی از ابزارهای پاییندستی مانند SAMtools و GATK تضمین میکند. به همین دلیل، Bowtie 2 اغلب به عنوان اولین گام در پایپلاینهای ژنومیکس مقایسهای شناخته میشود و کاربرد گستردهای در فرایندهایی نظیر فراخوانی واریانتها (Variation calling) ، ChIP-seq ، RNA-seq و BS-seq دارد.
این نرمافزار تحت لایسنس منبعباز GPLv3 منتشر شده و به راحتی از طریق خط فرمان (Command line) در سیستمعاملهای لینوکس، مک، ویندوز و BSD قابل اجراست.
برای درک بهتر جایگاه Bowtie 2 در بیوانفورماتیک مدرن، باید کمی به عقب برگردیم؛ به سال ۲۰۰۹، زمانی که فناوری توالییابی نسل جدید (NGS) با سرعتی باورنکردنی در حال تولید حجم عظیمی از دادهها بود. در آن دوران، بزرگترین چالش پژوهشگران، نگاشت و همترازی این حجم وسیع از خوانشهای کوتاه با ژنوم رفرنس، آن هم با منابع محاسباتی محدود و رایانههای معمولی بود.
در پاسخ به این چالش، Ben Langmead و همکارانش در دانشگاه Johns Hopkins، نسخه اول ابزار Bowtie (که امروزه به آن Bowtie 1 نیز میگویند) را معرفی کردند. این ابزار با بهرهگیری خلاقانه از الگوریتم BWT و ساختار داده FM-index، انقلابی در سرعت و مدیریت حافظه ایجاد کرد. Bowtie 1 قادر بود خوانشهای کوتاه آن زمان (حدود ۲۵ تا ۵۰ نوکلئوتید) را تا ۳۰ برابر سریعتر از ابزارهای همدوره خود الاین کند، در حالی که بخش بسیار کوچکی از حافظه رم را اشغال میکرد.
اما پیشرفت تکنولوژی توالییابی متوقف نشد. با گذشت زمان، دستگاههای توالییابی توانستند خوانشهای بلندتری (بیش از ۵۰ تا چند صد نوکلئوتید) تولید کنند. خوانشهای بلندتر، پتانسیل بیشتری برای مواجهه با جهشهای ساختاری مانند حذف و اضافهشدنهای کوچک (Indels) داشتند. اینجا بود که ضعف Bowtie 1 نمایان شد؛ این نسخه اولیه برای خوانشهای بسیار کوتاه بهینهسازی شده بود و توانایی انجام همترازی فاصلهدار (Gapped Alignment) برای تشخیص این تغییرات ساختاری را نداشت.
تیم توسعهدهنده برای رفع این محدودیت دست به کار شد و در سال ۲۰۱۲، ابزار Bowtie 2 را به جهان معرفی کرد. Bowtie 2 با بازطراحی الگوریتمها و افزودن قابلیتهای حیاتی مانند همترازی محلی (Local) و فاصلهدار، توانست با حساسیت و دقت بسیار بالاتر، خوانشهای بلندتر را همترازی کند. این ابزار بدون از دست دادن مزیتهای اصلی خود یعنی سرعت فوقالعاده و مصرف بهینه حافظه، به یکی از ارکان اصلی پایپلاینهای بیوانفورماتیکی مدرن تبدیل شد.
Bowtie 1 که در سال ۲۰۰۹ منتشر شد، برای خوانشهای بسیار کوتاه که در آن زمان رایج بود، بهینهسازی شده بود. اما Bowtie 2 برای همگامی با دیتای جدیدتر و طولانیتر توسعه یافت.
در ادامه، مهمترین تفاوتهای این دو ابزار را بررسی میکنیم:
۱. همترازی فاصلهدار (Gapped Alignment) و محلی (Local)
۲. سرعت و حساسیت بر اساس طول خوانش مهمترین معیار برای انتخاب بین این دو نسخه، طول خوانش دادههای شماست:
۳. سیستم امتیازدهی و انعطافپذیری
Bowtie 2 برای اینکه بتواند با سرعتی بینظیر میلیاردها خوانش را با ژنوم رفرنس مطابقت دهد، از یک فرآیند هوشمندانه چهار مرحلهای استفاده میکند. بر اساس مستندات منتشر شده در ژورنال Nature Methods، روند همترازی برای هر خوانش به شرح زیر است:
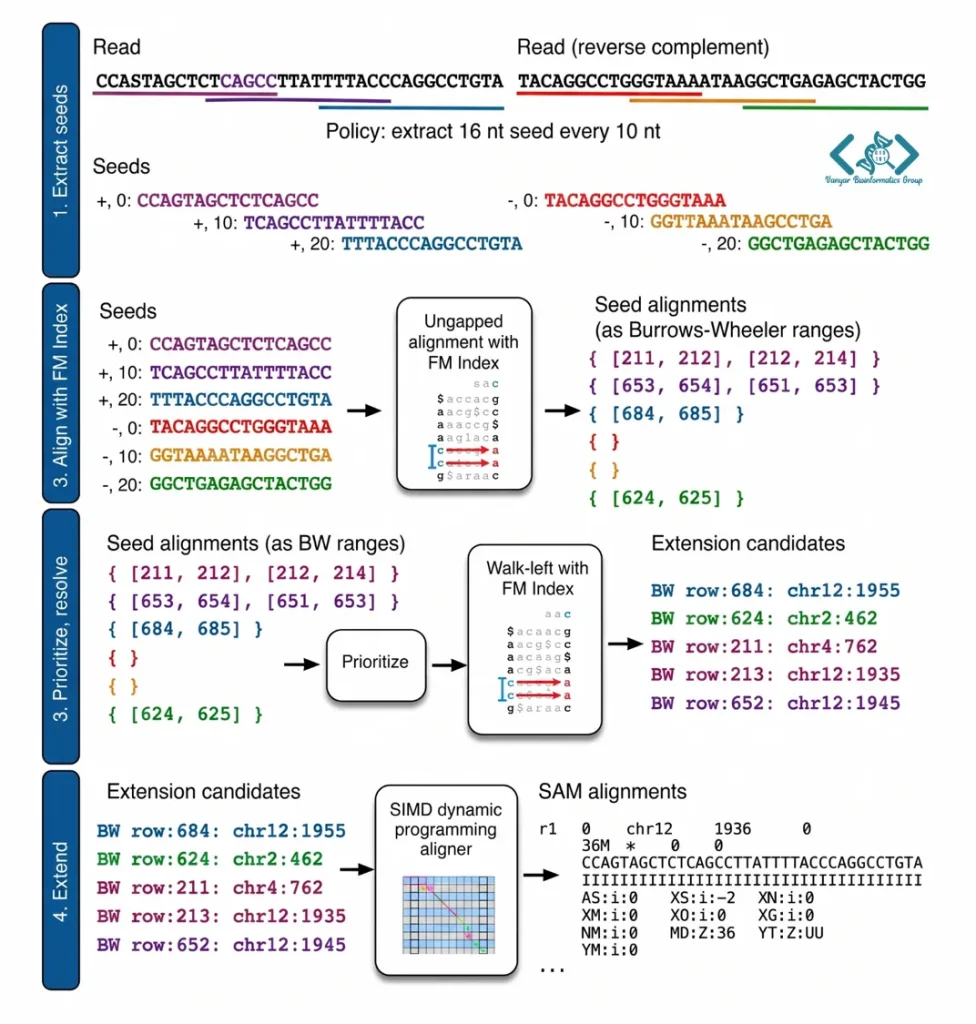
مرحله ۱: استخراج بذرها (Seed Extraction) : ابتدا الگوریتم، خوانش اصلی را به قطعات بسیار کوچکتری به نام «بذر» (Seed) تقسیم میکند. این عمل هم برای توالی اصلی و هم برای رشته مکمل معکوس (Reverse complement) آن انجام میشود تا هیچ احتمالی برای همترازی از دست نرود.
مرحله ۲: جستجوی اولیه با کمک FM Index : در این گام، Bowtie 2 بذرهای استخراجشده را به صورت بدون فاصله (Ungapped) با ژنوم مرجع مطابقت میدهد. این جستجوی فوقسریع به کمک ساختار داده FM Index انجام میشود و خروجی آن، بازههایی به نام محدودههای بروز-ویلر (BW Ranges) است که مکانهای احتمالی بذرها را روی ژنوم نشان میدهد.
مرحله ۳: اولویتبندی و موقعیتیابی دقیق (Walk-left) الگوریتم اکنون محدودههای BW بهدستآمده را بررسی میکند. در اینجا یک قانون مهم وجود دارد: محدودههای کوچکتر (که نشاندهنده تطابقهای اختصاصیتر هستند) اولویت بالاتری دریافت میکنند. سپس نرمافزار به صورت تصادفی اما بر اساس این اولویتها، موقعیت دقیق (Offset) هر بذر را روی ژنوم مرجع با استفاده از فرآیندی به نام “Walk-left” محاسبه و قطعی میکند.
مرحله ۴: همترازی نهایی با برنامهنویسی پویا (Dynamic Programming) در گام آخر، Bowtie 2 به سراغ موقعیتهای تاییدشده در مرحله قبل میرود و در مجاورت آنها، یک همترازی بسیار دقیق با روش «برنامهنویسی پویا» انجام میدهد. برای جلوگیری از افت سرعت، این محاسبات پیچیده توسط دستورات SIMD در پردازنده شتابدهی میشوند. این فرآیند جستجو تا زمانی ادامه مییابد که تمام بذرها بررسی شوند، تعداد کافی از الاینمنتها یافت شود، و یا اینکه الگوریتم به سقف مجاز محاسباتی خود برسد.
برای نصب Bowtie 2، بسته به سطح تخصص و نیاز شما، دو مسیر کلی وجود دارد: استفاده از نسخههای آماده یا نصب با سورس کد
این گزینهها برای کاربرانی است که میخواهند بدون درگیر شدن با کدهای برنامهنویسی، سریعاً کار الاینمنت را شروع کنند:
conda install bowtie2 در ترمینال، نرمافزار به طور کامل نصب میشود.رمز فایل فشرده: www.vanyarbioinf.ir
این روش مخصوص کاربرانی است که میخواهند نرمافزار را دقیقاً برای پردازنده سیستم خود بهینهسازی کنند.
در این حالت، شما فایل سورسکد را دانلود میکنید. این فایل شامل کدهای خام است و قابلیت اجرای مستقیم ندارد؛ بلکه باید آن را روی سیستم خودتان ترجمه (کامپایل) کنید:
make را اجرا کنید. این دستور، کدهای خام را به یک برنامه اجرایی تبدیل میکند.در زمان کامپایل سورسکد، توسعهدهندگان به شما اجازه میدهند ویژگیهای خاصی را فعال کنید:
make USE_SRA=1 انجام دهید.libsais برای ساخت ایندکس استفاده کنید. این روش سرعت ساخت ایندکس را به شدت افزایش میدهد، اما نسبت به حالت عادی، فضای RAM بسیار بیشتری اشغال میکند.بعد از اینکه نرمافزار را به هر یک از روشهای بالا آماده کردید، باید بتوانید دستورات آن را از هر پوشهای در سیستم اجرا کنید. حتماً مسیر پوشه حاوی فایلهای Bowtie 2 را به متغیر محیطی PATH در سیستمعامل خود اضافه کنید.
تصور کنید میخواهید یک کلمه خاص را در یک کتاب قطور پیدا کنید؛ اگر کتاب فهرست (Index) نداشته باشد، باید صفحه به صفحه آن را بگردید که کار بسیار زمانبری است. الگوریتمهای الاینمنت هم دقیقاً همین مشکل را با ژنومهای بزرگ (مثل ژنوم ۳ میلیارد جفتبازی انسان) دارند.
ابزار bowtie2-build فایلهای توالی ژنوم (FASTA) را دریافت کرده و با استفاده از الگوریتمهای پیشرفتهای نظیر تبدیل Burrows-Wheeler، یک ایندکس از آن میسازد. پس از ساخت این ایندکس، سرعت جستجو و الاینمنت به شدت افزایش مییابد.
bt2 دارند (مانند NAME.1.bt2 و NAME.rev.1.bt2)bt2l تغییر میکند. پس از تولید این ۶ فایل، نرمافزار Bowtie 2 برای انجام الاینمنت دیگر هیچ نیازی به فایل FASTA اولیه ندارد!برای اجرای این ابزار، ساختار کلی دستور به این شکل است:
bowtie2-build [options] <reference_in> <bt2_base>در میان دهها تنظیم مختلف، این موارد برای کارهای روزمره از همه مهمترند:
<reference_in>: مسیر فایلهای FASTA ژنوم مرجع (میتواند چند فایل باشد که با کاما جدا شدهاند)<bt2_base>: نام دلخواهی که میخواهید برای فایلهای خروجی ایندکس انتخاب کنید.--threads: این گزینه حیاتی است! به صورت پیشفرض برنامه فقط از یک هسته پردازنده استفاده میکند. با افزایش این عدد، سرعت ساخت ایندکس به شدت بالا میرود.--large-index: اگر ژنوم شما زیر ۴ میلیارد باز است اما همچنان اصرار دارید ساختار ایندکس از نوع بزرگ (Large index) باشد، این گزینه را فعال کنید.نکته مهم : ساخت ایندکس برای ژنومهای بزرگی مثل ژنوم انسان بسیار زمانبر است و سیستم شما را به شدت درگیر میکند. خبر خوب این است که نیازی نیست همیشه ایندکسها را از صفر بسازید! توسعهدهندگان Bowtie 2 و مراجعی مانند پایگاه Illumina iGenomes، ایندکسهای آماده و استانداردی را برای مهمترین ارگانیسمها (مانند انسان، موش، مگس سرکه و مخمر) از پیش محاسبه کرده و برای دانلود رایگان قرار دادهاند.
ساختار کلی و استاندارد برای اجرای نرمافزار Bowtie 2 در خط فرمان به شکل زیر است:
bowtie2 [options] -x <bt2-idx> {-1 <m1> -2 <m2> | -U <r>} -S [<sam>]
در نگاه اول شاید این ساختار کمی گیجکننده به نظر برسد، اما در واقع از چند بخش بسیار ساده و منطقی تشکیل شده است. بیایید آرگومانهای اصلی (Main Arguments) را رمزگشایی کنیم:
-x (ایندکس ژنوم مرجع): این مهمترین آرگومان است. در اینجا باید مسیر و «نام پایه» فایلهای ایندکسی را که در مرحله قبل (با دستور bowtie2-build) ساختهاید، وارد کنید.
.1.bt2 را بنویسید؛ فقط همان نام اصلی کافی است.-1 و -2 (برای دادههای Paired-end): اگر دادههای توالییابی شما جفتانتهایی هستند، فایلهای خوانش اول (Forward) را با آرگومان -1 و خوانشهای دوم (Reverse) را با آرگومان -2 به برنامه میدهیم (این فایلها معمولاً فرمت FASTQ دارند).-U (برای دادههای Single-end): اگر دادههای شما تکانتهایی (Unpaired) هستند، به جای استفاده از دو آرگومان بالا، تنها از -U استفاده کرده و مسیر فایل خوانش خود را وارد میکنید.-S (فایل خروجی): در این بخش نام و مسیر فایلی را که میخواهید نتایج الاینمنت در آن ذخیره شود، مشخص میکنید. این فایل به صورت استاندارد باید پسوند .sam داشته باشد.(سایر ورودیهای خاص): علاوه بر موارد بالا، Bowtie 2 میتواند به صورت مستقیم دادههای دانلودشده از پایگاه NCBI (با پارامتر --sra-acc) یا فایلهای الایننشده BAM (با پارامتر -b) را نیز به عنوان ورودی دریافت کند.
توسعهدهندگان Bowtie 2، دیتای آموزشی ویروس باکتریوفاژ لامبدا را درون پوشه نرمافزار قرار دادهاند تا بتوانید کل مسیر الاینمنت تا فراخوانی واریانتها (variant calling) را در چند دقیقه تمرین کنید.
برای اجرای دستورات در ترمینال لینوکس، فرض میکنیم متغیر محیطی BT2_HOME را به پوشه نصب Bowtie 2 متصل کردهاید.
ابتدا باید ژنوم مرجع ویروس لامبدا (فایل FASTA) را برای نرمافزار قابلفهم کنیم. یک پوشه جدید بسازید و دستور زیر را اجرا کنید:
$BT2_HOME/bowtie2-build $BT2_HOME/example/reference/lambda_virus.fa lambda_virus
.bt2 میسازد که همان ایندکسهای ژنوم مرجع شما هستند.حالا با توجه به نوع دادههایتان، یکی از دستورات زیر را برای همترازی اجرا کنید. خروجی همه این حالتها، یک فایل متنی با فرمت استاندارد SAM خواهد بود که حاوی موقعیت دقیق خوانشها روی ژنوم است.
$BT2_HOME/bowtie2 -x lambda_virus -U $BT2_HOME/example/reads/reads_1.fq -S eg1.sam
$BT2_HOME/bowtie2 -x lambda_virus -1 $BT2_HOME/example/reads/reads_1.fq -2 $BT2_HOME/example/reads/reads_2.fq -S eg2.sam
$BT2_HOME/bowtie2 --local -x lambda_virus -U $BT2_HOME/example/reads/longreads.fq -S eg3.sam
فایل SAM متنی و بسیار حجیم است. در پایپلاینهای واقعی، ما از ابزارهای SAMtools و BCFtools برای فشردهسازی، مرتبسازی و در نهایت یافتن واریانتها استفاده میکنیم. این فرآیند سه مرحله دارد:
تبدیل SAM به BAM (فشردهسازی باینری) – مرتبسازی فایل BAM (الزامی برای مراحل بعدی – استخراج واریانتها (تبدیل به VCF)
در دنیای آنالیز دادههای ژنومی، هیچ ابزاری شبیه یک آچار فرانسه نیست که برای تمام پروژهها بینقص کار کند. پژوهشگران بارها Bowtie 2 را در کنار دهها ابزار دیگر (مانند BWA ، GSNAP ، SOAP2 و غیره) در شرایط واقعی آزمایش کردهاند تا ببینند این نرمافزار دقیقاً در چه مواردی میدرخشد و کجا باید جای خود را به ابزارهای دیگر بدهد.
بیایید جایگاه Bowtie 2 را در برابر رقبای اصلیاش در پروژههای مختلف بررسی کنیم:
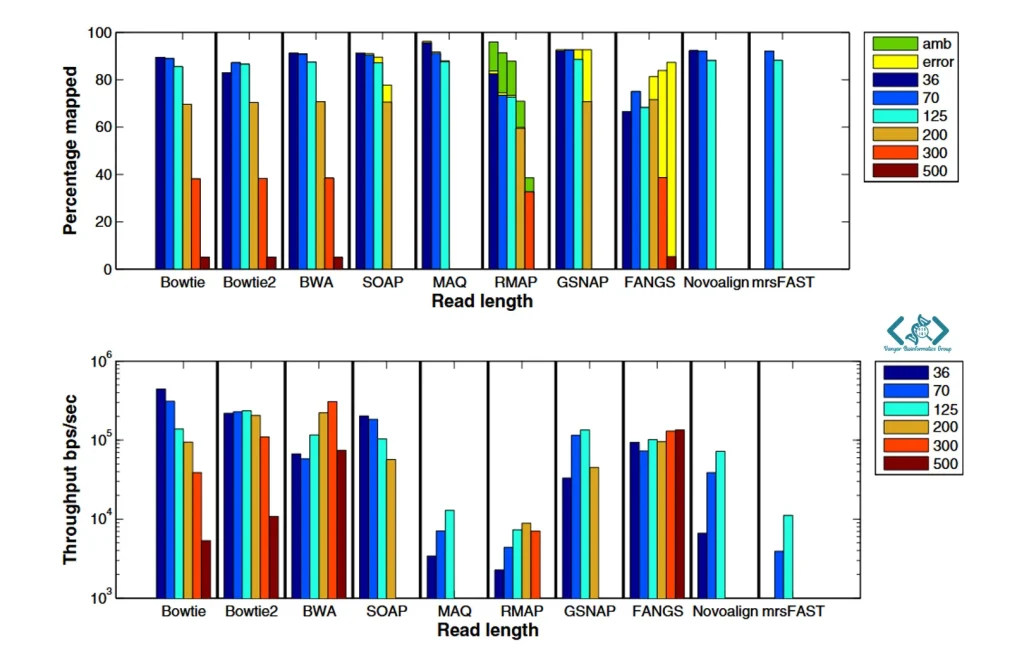
بسیاری از نرمافزارهای قدیمیتر با افزایش طول خوانشهای توالییابی دچار افت شدید سرعت و دقت میشدند. اما بررسیها نشان میدهد که Bowtie 2 (در کنار BWA) از معدود ابزارهایی است که میتواند خوانشهای بلند (حتی تا ۵۰۰ جفتباز) را با پایداری بسیار بالایی همتراز کند. نکته جالب اینجاست که Bowtie 2 در تنظیمات پیشفرض خود، سرعت را به بالاترین حدِ ممکن میرساند؛ به همین دلیل ممکن است درصد اندکی از خوانشهای پیچیده را از دست بدهد. اما جای نگرانی نیست! اگر در پروژهای نیاز به دقتِ وسواسگونه دارید، به راحتی میتوانید با تغییر چند پارامتر، حساسیت نرمافزار را بالا ببرید (هرچند این کار کمی زمان اجرا را طولانیتر میکند).
وقتی پای استخراج واریانتهای ژنتیکی (SNP Calling) به میان میآید، Bowtie 2 عملکرد بسیار قابلقبولی دارد و در آزمایشها میتواند هزاران واریانت را با دقت بالا استخراج کند. با این حال، رقیب اصلی او یعنی BWA-MEM در این زمینه پادشاهی میکند!
اینجا دقیقاً همان جایی است که باید در انتخاب ابزار دقت کنید! دادههای RNA-Seq حاوی اطلاعات اگزونها هستند و نرمافزار باید بتواند فواصل بزرگِ اینترونی روی ژنوم را تشخیص دهد (به این ویژگی Splice-awareness میگویند). Bowtie 2 این قابلیت را ندارد؛ بنابراین برای همترازی RNA با ژنوم مرجعِ یوکاریوتها مناسب نیست (مگر اینکه روی باکتریها کار کنید یا مرجع شما ترانسکریپتوم باشد).
تسلط بر ابزارهای پایهای و قدرتمندی مانند Bowtie 2، سنگ بنای موفقیت در پروژههای پیچیده بیوانفورماتیکی است. درک عمیق از نحوه عملکرد الگوریتمها، مدیریت بهینه منابع سیستم و انتخاب هوشمندانه ابزار الاینمنت (با آگاهی از نقاط ضعف و قوت آن در برابر رقبایی مثل BWA-MEM یا HISAT2)، دقیقاً همان چیزی است که تفاوت یک آنالیز معمولی و یک خروجی حرفهای و قابلاتکا را رقم میزند.
نرمافزار Bowtie 2 با ترکیب بینظیر سرعت فوقالعاده، مصرف بسیار پایین حافظه (RAM) و پایداری بالا، همچنان یکی از مطمئنترین سلاحها در جعبهابزار هر پژوهشگر است. با اجرای قدمبهقدم مراحل نصب، ساخت ایندکس و اجرای الاینمنت که در این راهنما بررسی کردیم، اکنون شما پلتفرم مستحکمی در اختیار دارید تا با اطمینان کامل دیتای خام توالییابی را پردازش کرده و مسیر را برای کشفیات بزرگ ژنومی خود هموار کنید.
۱. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature methods. 2012 Apr;9(4):357-9
۲. Hatem A, Bozdağ D, Toland AE, Çatalyürek ÜV. Benchmarking short sequence mapping tools. BMC bioinformatics. 2013 Jun 7;14(1):184.
Bowtie 2 یک ابزار همترازی (Aligner) فوقسریع و کممصرف است که وظیفه نگاشت (Mapping) میلیونها خوانش کوتاه توالییابی به یک ژنوم مرجع بزرگ را بر عهده دارد و پیشنیاز اصلی در اکثر آنالیزهای ژنومیکس محسوب میشود.
Bowtie 2 پادشاه بلامنازع دادههای DNA (مانند ChIP-Seq ، BS-Seq و WGS) است. اما برای دادههای RNA-Seq یوکاریوتها مناسب نیست (زیرا فواصل اینترونی را تشخیص نمیدهد)، مگر اینکه بخواهید دادهها را با یک «ترانسکریپتوم» (بدون اینترون) یا ژنوم باکتریها همتراز کنید.
بله، Bowtie 2 یک نرمافزار منبعباز (Open-source) و ۱۰۰٪ رایگان است که تحت لایسنس بینالمللی GPLv3 منتشر شده و تمامی پژوهشگران میتوانند بدون هیچگونه محدودیتی آن را روی لینوکس، مک و ویندوز نصب و استفاده کنند.
نسخه اول (Bowtie 1) منحصراً برای خوانشهای بسیار کوتاه (زیر ۵۰ جفتباز) ساخته شده بود. اما Bowtie 2 برای خوانشهای بلندتر بهینهسازی شده و از همترازی فاصلهدار (Gapped) و محلی (Local) برای یافتن واریانتها پشتیبانی میکند.
بله، Bowtie 2 به طور کامل هم برای دادههای تکانتهایی (Single-end) و هم جفتانتهایی طراحی شده است. جالبتر اینکه اگر نتواند دو خوانش را به صورت جفت همتراز کند، تلاش میکند هر کدام را جداگانه (Unpaired) مطابقت دهد.

تیم تولید محتوای گروه بیوانفورماتیک وانیار در تلاش است تا بهترین آموزشهای کوتاه در زمینه بیوانفورماتیک و زیستشناسی را تهیه نماید. صحت محتوای این صفحه توسط کارشناسان گروه بیوانفورماتیک وانیار بررسی شده است.




عضویت در مجله وانیار
چطور از جدیدترین آموزشها باخبر شوم؟
با عضویت در مجله بیوانفورماتیک وانیار، برترین آموزشهای بیوانفورماتیک را در لحظه انتشار دریافت کنید.
سلام، وقت بخیر.
چطور میتونیم بهتون کمک کنیم؟
تیم ما آماده پاسخگویی به سوالات شماست.
پشتیبانی 24 ساعته در 7 روز هفته.