در عصر حاضر، فناوریهای توالییابی نسل جدید (NGS) حجم عظیمی از دادههای بیولوژیکی را با سرعتی بیسابقه تولید میکنند. درک و تفسیر این کوهِ دیتا، بدون ابزارهای بصری مناسب تقریباً غیرممکن است. اینجاست که Integrative Genomics Viewer یا به اختصار IGV وارد میدان میشود.
نرمافزار IGV که توسط Broad Institute طراحی و توسعه پیدا کرده، یکی از قدرتمندترین، محبوبترین و پرکاربردترین ابزارهای رایگان و متنباز برای تجسم تعاملی دادههای ژنومیک به شمار میآید. IGV در کنار روشهای تحلیل متنی و خروجیهای عددی، با ارائه یک نمایش گرافیکی یکپارچه، الگوهای پنهان در دادهها را آشکار میسازد و به کاربران اجازه میدهد تا مناطق مورد علاقه را به سرعت بررسی کنند.
با استفاده از این ابزار، پژوهشگران میتوانند طیف وسیعی از دادههای ژنومیک (از فایلهای الاینمنت مانند BAM و SAM گرفته تا فایلهای واریانت مثل VCF و فایلهای تفسیری BED) را به صورت لایهبندی شده و همزمان مشاهده کنند. رابط کاربری هوشمند IGV به شما اجازه میدهد تا با یک اسکرول ساده، از نمای کلی یک کروموزوم تا سطح تکتک نوکلئوتیدها زوم کنید و مناطق مورد علاقه خود را با دقتی بینظیر مورد بررسی قرار دهید.
IGV فراتر از یک نرمافزار معمولی، یک پلتفرم جامع و چندمنظوره است که با ارائه ابزارها و نسخههای متنوع، نیازهای طیف گستردهای از کاربران—از پژوهشگران تا توسعهدهندگان وب—را به بهترین شکل برآورده میکند. نکته بسیار جذاب برای توسعهدهندگان این است که تمامی نرمافزارهای این پلتفرم کاملاً متنباز (Open Source) بوده و تحت لایسنس MIT در مخازن GitHub در دسترس هستند.
این پلتفرم قدرتمند شامل نسخهها وابزارهای زیر است:
نسخه دسکتاپ، کاملترین محیط کاری IGV برای آنالیز سریع و دقیق فایلهای حجیم توالییابی (مانند فایلهای BAM کل ژنوم یا WES) است.
از نسخه 2.19.1 به بعد، این نرمافزار به Java 21 نیاز دارد. برای جلوگیری از مشکلات نصب پیشنیازها، توسعهدهندگان دو نوع پکیج دانلودی ارائه میدهند:
رمز فایل فشرده: www.vanyarbioinf.ir
نسخه خط فرمان (Command line) نرمافزار IGV و igvtools مخصوص افرادی است که میخواهند از طریق ترمینال و اسکریپتنویسی، کارهای سنگین و خودکار را روی سرورها انجام دهند. این نسخه برای تمامی پلتفرمها از لینک زیر قابل دانلود است:
رمز فایل فشرده: www.vanyarbioinf.ir
برای شروع، بیایید مراحل نصب روی سیستمعامل ویندوز را به صورت گامبهگام مرور کنیم:
.exe) دو بار کلیک کنید تا پنجره نصب باز شود.اگر نیازی به نصب نرمافزار ندارید یا میخواهید یک بررسی سریع روی دیتا داشته باشید، نسخه تحت وب IGV نیاز شما را برطرف میکند. این نسخه مستقیماً از طریق مرورگر در دسترس است و برای اشتراکگذاری سریع نتایج با همکاران ایدهآل است.
این نسخه یک کتابخانه قدرتمند جاوا اسکریپت است. هدف از خلق igv.js این بود که توسعهدهندگان بتوانند اجزای تعاملی مصورسازی ژنوم را بدون نیاز به نصب نرمافزار جانبی، مستقیماً درون برنامههای تحت وب خود ادغام کنند. برای درک اهمیت این نسخه، جالب است بدانید که پلتفرمهای بزرگ و معروفی مانند cBioPortal که برای مصورسازی دادههای سرطان استفاده میشوند، از همین کتابخانه برای نمایش دیتای خود بهره میبرند که نشاندهنده انعطافپذیری فوقالعاده آن است.
این نسخه در واقع یک رابط یکپارچه پایتون برای igv.js است که مخصوص محیطهای تعاملی طراحی شده است. این پکیج به محققان اجازه میدهد تا بدون نیاز به خروج از نوتبوکهایی مثل Jupyter یا Google Colab، مصورسازیهای ژنومی خود را به راحتی اجرا و بررسی کنند.
ابزاری فوقالعاده برای تولید فایلهای مستقل HTML است که نتایج کار شما (شامل مناطق ژنومی و تصاویر IGV) را در یک جا جمعآوری میکند. با این کار، اشتراکگذاری نتایج با همکاران و اساتید، سریعتر و حرفهایتر از همیشه انجام میشود.
در واقع، توسعهدهندگان IGV تنها یک مرورگر ژنومی طراحی نکردهاند؛ آنها اکوسیستمی کامل از ابزارهای دسکتاپ، تحت وب و توسعهای را در اختیار کاربران قرار دادهاند. این رویکرد باعث شده است پژوهشگران، متخصصان ژنتیک، دانشمندان دیتا و توسعهدهندگان وب بتوانند از قابلیتهای مصورسازی ژنومیک IGV در طیف گستردهای از پروژهها استفاده کنند.
پس از اجرای IGV، با یک محیط کاری یکپارچه مواجه میشوید که از چند بخش کلیدی برای مدیریت و نمایش دادهها تشکیل شده است:
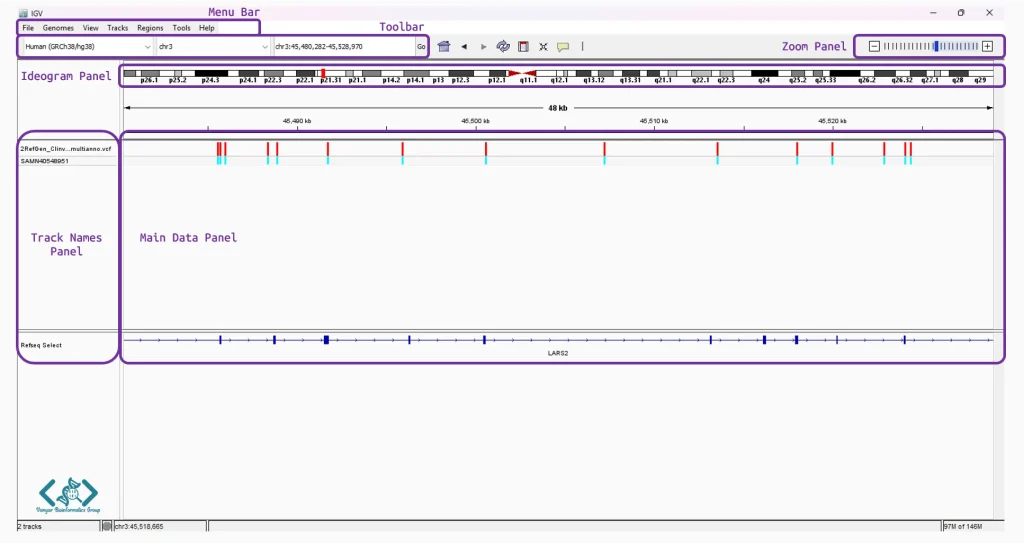
کنترل همهجانبهی نرمافزار را در اختیار شما میگذارد. در این نوار، منوی File برای فراخوانی دادهها، ذخیره فضاهای کاری و خروجی گرفتن از تصاویر کاربرد دارد. منوی Genomes به طور تخصصی برای بارگذاری ژنومهای مرجع استفاده میشود. منوهای View و Tracks به شما اجازه میدهند تا ظاهر محیط کار و نحوه نمایش، گروهبندی یا فیلتر کردن لایههای داده را کاملاً شخصیسازی کنید. همچنین از طریق منوی Regions میتوانید مناطق حساس و مهم ژنومی را نشانهگذاری کنید و منوی Tools نیز دسترسی سریع به ابزارهای پردازشی قدرتمندی مانند igvtools را برای شما فراهم میکند.
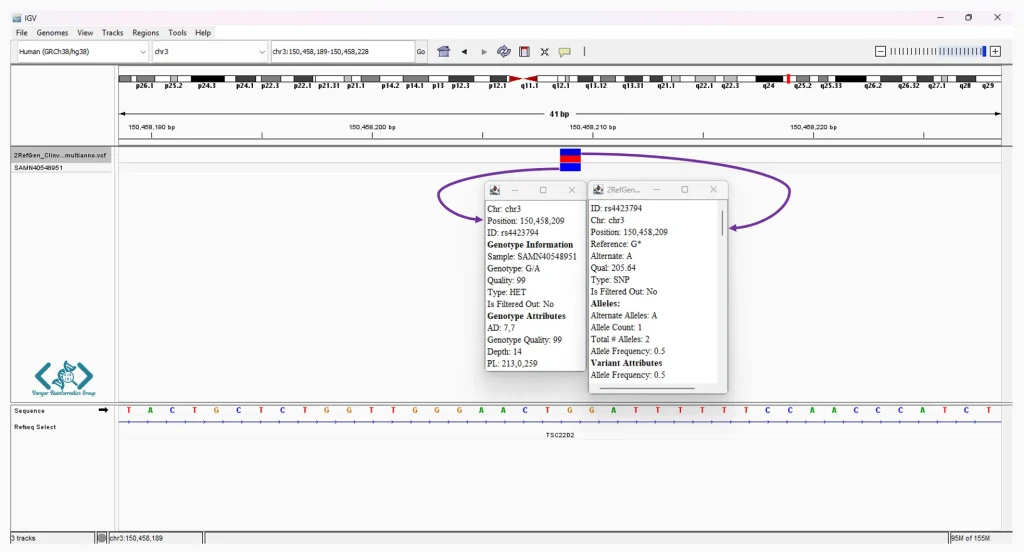
دسترسی سریع به تنظیمات اصلی را فراهم میکند. همانطور که در تصویر مشخص است، در سمت چپ این نوار، منوهای کشویی برای انتخاب سریع ژنوم مرجع (مانند Human (GRCh38/hg38)) و انتخاب کروموزوم (مانند chr3) قرار دارند. در کنار آنها، نوار جستجو تعبیه شده است که به شما اجازه میدهد با وارد کردن مختصات دقیق یا نام ژنها، مستقیماً به منطقه مورد نظر پرش کنید. ابزارهای بزرگنمایی نیز در سمت راستِ همین نوار قرار گرفتهاند.
برای شروع تحلیل، اولین قدم تعیین ژنوم مرجع است. میتوانید مستقیماً از اولین منوی کشویی در سمت چپ نوار ابزار (که در این تصویر روی
Human (GRCh38/hg38)تنظیم شده است)، ژنومهای پیشفرضِ ابری را انتخاب کنید. اگر روی ارگانیسم خاصی کار میکنید که در لیست پیشفرض نیست، این انعطافپذیری وجود دارد که از طریق منویGenomesدر نوار بالای نرمافزار، فایلهای توالی (FASTA) و ایندکس محلی خود را به راحتی به محیط کار اضافه کنید.
ویژگی بسیار کاربردی این بخش، وجود یک نشانگر یا نوار قرمز رنگ کوچک است که دقیقاً مشخص میکند محدودهای که در حال حاضر در پنل پایین مشاهده میکنید، در کجای کل کروموزوم قرار دارد.
نام فایلها و دادههای بارگذاریشده را نشان میدهد (مانند فایل واریانت .vcf یا مسیر مرجع Refseq Select). این پنل به شما کمک میکند تا بدانید هر ردیف از دادهها مربوط به کدام نمونه یا فایل است.
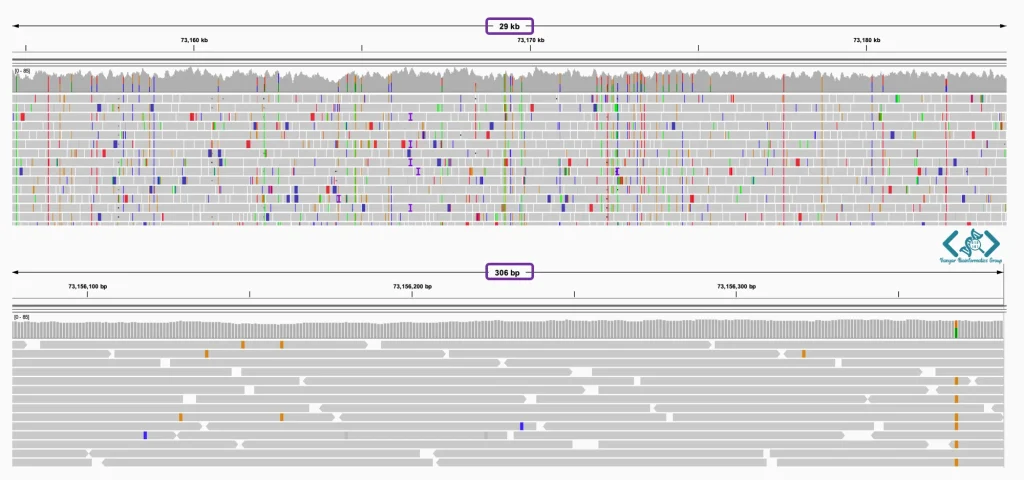
در این قسمت، دادههای ژنومیک به صورت لایههای افقی (Tracks) روی هم قرار میگیرند. برای مثال در این نما، لاینهای رنگی عمودی (قرمز و آبی) نشاندهنده موقعیت واریانتها هستند و در پایینترین لایه، حاشیهنویسی ژنوم (مانند ژن LARS2) به عنوان یک نقشه راه برای تفسیر دادهها نمایش داده شده است.
وقتی دادهها به درستی در IGV بارگذاری شدند، قدرت واقعی این نرمافزار در کشف حقایق پنهان ژنوم خود را نشان میدهد. IGV صرفاً یک ابزار نمایش نیست، بلکه یک پلتفرم تحلیلی است که به محققان اجازه میدهد فرضیههای خود را به صورت بصری ارزیابی کنند. در ادامه، مهمترین کاربردهای تحلیلی این نرمافزار را بررسی میکنیم:
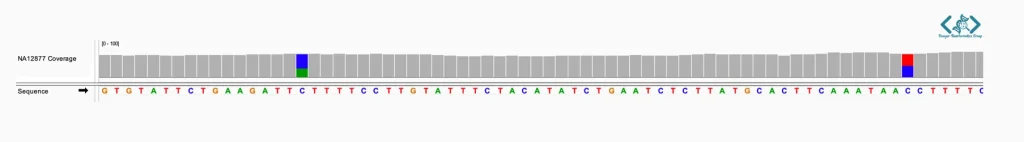
شناسایی واریانتها (SNPs و InDels) معمولاً توسط نرمافزارهای الگوریتمی (Variant Callers) انجام میشود، اما تایید نهایی آنها نیاز به بررسی چشمی دارد. IGV ابزاری قدرتمند برای تایید و بررسی واریانتهای شناساییشده توسط نرمافزارهای فراخوانی واریانت است. IGV با نمایش دقیق خوانشها در مقایسه با ژنوم مرجع، امکان اعتبارسنجی بصری واریانتها را فراهم میکند. این قابلیت به پژوهشگران اجازه میدهد خطاهای همترازی، نواحی کمپوشش و مناطق پیچیده یا تکراری ژنوم را با دقت بیشتری شناسایی و بررسی کنند. در این محیط، هر نوکلئوتیدی که با ژنوم مرجع مغایرت داشته باشد، با رنگهای مشخصی برجسته میشود که تشخیص جهشها را در یک نگاه ممکن میسازد.
فراتر از جهشهای نقطهای، نرمافزار IGV ابزاری قدرتمند برای بررسی تغییرات ساختاری وسیع (SVs) مانند حذفها (Deletions)، جابجاییها (Translocations)، معکوسشدگیها (Inversions) و مضاعفشدگیها (Duplications) است.
در دادههای همترازی (فایلهای BAM)، این تغییرات ساختاری معمولاً از طریق دو نشانه بصری مهم در IGV قابل شناسایی هستند:
۱. الگوی ناهنجار جفتخوانشها (Discordant Read Pairs): در توالییابی Paired-end، انتظار میرود جفتخوانشها فاصله و جهتگیری استانداردی روی ژنوم مرجع داشته باشند. IGV هرگونه انحراف از این الگو را با تغییر رنگ خوانشها به شما هشدار میدهد. به عنوان مثال:
۲. خوانشهای دوپاره (Split Reads / Soft-clipping): در حالی که تغییر رنگ جفتخوانشها وجود یک تغییر ساختاری را به ما خبر میدهد، برای پیدا کردن دقیقِ نقطه شکست (Breakpoint) باید به دنبال خوانشهایی باشیم که دقیقاً روی مرز این تغییر قرار گرفتهاند. در چنین مواردی، تنها بخشی از خوانش با ژنوم مرجع همتراز میشود. IGV بخش باقیمانده را به شکل حروف رنگی و Soft-clipped در انتهای خوانش نشان میدهد.
مثال بالینی: بررسی ادغامهای ژنی (Gene Fusions) یکی از کاربردهای جذاب این قابلیت، تشخیص ادغام ژنهای BCR و ABL1 در سرطان لوسمی میلوئیدی مزمن (CML) است. محقق با مراجعه به این نواحی در IGV، ابتدا جفتخوانشهایی را میبیند که یک سرِ آنها در کروموزوم ۹ و سرِ دیگر در کروموزوم ۲۲ مپ شده است. سپس با زوم کردن و بررسی دقیقِ Split Readها در همان ناحیه، میتواند نقطه دقیقِ شکست کروموزومی و اتصال این دو ژن را با وضوح بالا تایید کند.
دادههای ترانسکریپتومیک و RNA-seq داستان متفاوتی دارند. در اینجا، IGV نرمافزاری ایدهآل برای تجسم پوشش (Coverage) در مناطق مختلف ژن است که این سطح پوشش، مستقیماً نشاندهنده میزان بیان آن ژن است. محققان میتوانند با مقایسه ارتفاع نمودارهای پوشش در نمونههای بیمار و سالم، افزایش یا کاهش بیان ژنها را بررسی کنند. علاوه بر این، میتوان الگوهای پیچیدهای مانند پیرایش متناوب (Alternative Splicing) را با مشاهده دقیق خوانشهایی که از محل پیوندگاههای اگزون-اینترون عبور میکنند، شناسایی کرد.
برای محققانی که روی تنظیم بیان ژن کار میکنند، IGV برای تجسم دادههای ChIP-seq که در مطالعات اپیژنتیک استفاده میشوند، بسیار مناسب است. این نرمافزار توزیع سیگنالهای ChIP-seq را در طول ژنوم به صورت پیک نشان میدهد و به محققان اجازه میدهد تا نواحی غنیشده از هیستونهای اصلاحشده یا فاکتورهای رونویسی را کشف کنند.
موسسه Broad ابزار igvtools را برای پیشپردازش و بهینهسازی دادههای ژنومی توسعه داده است تا کاربران بتوانند فایلهای خود را سریعتر و کارآمدتر در IGV مشاهده کنند. این ابزار به عنوان یک دستیار برای پیشپردازش دادهها عمل میکند. زمانی که محققان با فایلهای ژنومیک بسیار حجیم (مانند فایلهای BAM) سروکار دارند، بارگذاری مستقیم آنها در نرمافزار ممکن است باعث کندی شدید سیستم یا پر شدن حافظه شود.
ابزار igvtools برای حل این مشکل ایجاد شده و با انجام کارهای زیر روی فایلهای خام، آنها را برای نمایش سریع و بهینه در IGV آماده میکند:
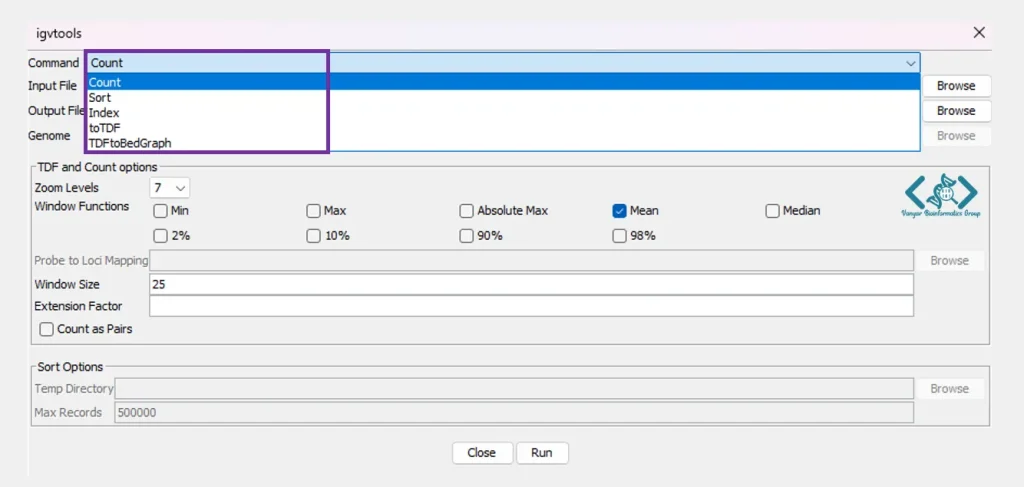
پیش از انجام هرگونه پیشپردازشی، دادههای ژنومیک باید بر اساس موقعیت کروموزومیشان مرتب شوند. ابزار Sort در igvtools، فایلهای شما (مانند BAM ، VCF یا BED) را میخواند و آنها را دقیقاً بر اساس مختصات شروع سازماندهی میکند. این کار پیشنیاز اصلی برای استفاده از سایر ابزارهای این مجموعه است.
همانطور که برای پیدا کردن یک کلمه در یک کتاب قطور به فهرست کلمات مراجعه میکنید، IGV نیز برای پیدا کردن یک ژن خاص در میان میلیاردها خوانش، به فایل Index نیاز دارد. این ابزار فایلهای مرتبشده را بررسی کرده و فایلهای Index (با پسوندهای .sai یا .idx) را در کنار آنها تولید میکند.
نکته جالب اینجاست که نیازی نیست این فایلهای Index را دستی بارگذاری کنید؛ همین که فایل اصلی را در IGV باز کنید، نرمافزار بهطور هوشمند فایل Index مجاور آن را شناسایی کرده و سرعت پیمایش را هزاران برابر میکند.
وقتی با دادههای پیوسته و بسیار حجیم کار میکنید، بهترین راهکار استفاده از ابزار toTDF است (که قبلاً با نام Tile شناخته میشد). این ابزار دادههای خام شما را به فرمت فوقفشرده و چندرزولوشنی .tdf تبدیل میکند. با این کار، زمانی که از نمای دور به یک کروموزوم نگاه میکنید، IGV تنها یک نمای کلی و سبک از دادهها را بارگذاری میکند و هرچه بیشتر زوم کنید، جزئیات بیشتری را فراخوانی میکند که نتیجه آن تجربهای کاملاً روان و بدون لگ است.
این ابزار به طور ویژه برای تحلیل پوشش خوانشها (Coverage) در فایلهای همترازی یا دادههای ChIP-seq طراحی شده است. ابزار Count با حرکت در طول ژنوم (در بازههای پنجرهای مشخص، مثلاً ۲۵ جفتباز)، میانگین تراکم دادهها را محاسبه کرده و خروجی آن را در قالب فرمت .tdf یا .wig ذخیره میکند. وقتی این فایل را در IGV بارگذاری میکنید، به جای دیدن میلیونها خوانش متراکم، یک نمودار میلهای (Bar Chart) بسیار واضح و خوانا از میزان پوشش هر منطقه خواهید دید که تحلیل الگوها را بسیار ساده میکند.
در ادامه مقاله، مهمترین فرمتهای قابل پشتیبانی در IGV را به دستههای کاربردی تقسیم کردهایم تا به راحتی بتوانید دادههای پایپلاینهای خود را وارد این نرمافزار کنید.
این فایلها حاصل مپ کردن خوانشها روی ژنوم مرجع هستند و هسته اصلی بررسیهای کیفی در آنالیز دادههای NGS محسوب میشوند.
.bai داشته باشد و کنار فایل اصلی قرار گیرد).Picard یا samtools) نیاز دارید..crai نیاز دارد..bam.list ذخیره کنید.پس از همترازی، نوبت به شناسایی واریانتها میرسد.
igvtools یا Tabix ایندکس کنید.برای اینکه بدانیم واریانتها در کجای ژنها رخ دادهاند، به فایلهای حاشیهنویسی نیاز داریم.
گاهی نیاز داریم توالیها را نه به صورت نقطهای، بلکه به عنوان یک نمودار پیوسته (مثل میزان بیان ژن یا عمق خوانش) ببینیم.
igvtools (دستور toTDF) ساخته میشود و سرعت لود دادههای گرافیکی را به شدت افزایش میدهد.انتخاب مرورگر ژنومی مناسب کاملاً به معماری دادهها و هدف پژوهش شما بستگی دارد. در دنیای بیوانفورماتیک، ابزار IGV به عنوان قهرمان بررسیهای محلی (Desktop-based) شناخته میشود. نقطه قوت بیرقیب IGV، پردازش سریع فایلهای حجیم همترازی (مانند BAM و CRAM) روی سیستم شخصی و اعتبارسنجی چشمی واریانتها در سطح تکتک خوانشها است. با این حال، به دلیل ماهیت آفلاینش، برای مقایسههای وسیع با دیتابیسهای آنلاین ایدهآل نیست.
در مقابل، UCSC Genome Browser یک پلتفرم قدرتمند مبتنی بر وب و معدن طلای حاشیهنویسیها (annotations) است. اگر پروژه شما نیازمند بررسی ژنها در بستر عناصر تنظیمی، مسیرهای حفاظتشده تکاملی و دادههای پروژه ENCODE باشد، UCSC بهترین انتخاب است. با اینحال، رابط کاربری نسبتاً قدیمی آن و چالشهای آپلود فایلهای حجیم شخصی (بهصورت Track Hub) میتواند برای کاربران تازهکار کمی کند و زمانبر باشد.
Ensembl یک پلتفرم تحت وب با تمرکز بر ژنومیک تطبیقی است. محققان از این ابزار برای شناسایی ارتولوگها، بررسی درختهای فیلوژنتیک و پیشبینی اثر واریانتها با کمک ابزار VEP استفاده میکنند. Ensembl اطلاعات ژنومی بسیار گستردهای را در اختیار کاربران قرار میدهد، اما همین گستردگی اطلاعات و امکانات ممکن است یادگیری و استفاده از آن را برای کاربران مبتدی کمی چالشبرانگیز کند.
در نهایت، JBrowse (بهویژه JBrowse 2) نسل جدید و انتخاب مدرن تیمهای تحقیقاتی است. این ابزار با رابط کاربری روان و تمرکز ویژه بر پشتیبانی بومی از خوانشهای بلند (Long-reads مانند ONT و PacBio)، واریانتهای ساختاری و نمایش سینتنی (Synteny) میدرخشد. برخلاف UCSC و Ensembl که دادهها و حاشیهنویسیهای آماده ارائه میکنند، JBrowse یک پلتفرم منعطف برای بارگذاری و مدیریت دادههای اختصاصی است. به همین دلیل بسیاری از تیمهای تحقیقاتی از آن برای ساخت پورتالهای ژنومیک اختصاصی روی سرورهای خود استفاده میکنند.
Integrative Genomics Viewer (IGV) یکی از قدرتمندترین ابزارهای مصورسازی و تحلیل دادههای ژنومیک است که پژوهشگران با کمک آن دادههای پیچیده توالییابی را به شکلی بصری، تعاملی و قابل فهم بررسی میکنند. این نرمافزار با پشتیبانی از انواع دادههای ژنومی، قابلیتهای گسترده سفارشیسازی و ابزارهای تحلیلی متنوع، جایگاه ویژهای در پژوهشهای بیوانفورماتیک، ژنتیک و ژنومیک به دست آورده است.
پژوهشگران از IGV برای طیف گستردهای از کاربردها، از جمله بررسی واریانتهای ژنتیکی، تحلیل تغییرات ساختاری ژنوم، مطالعه الگوهای بیان ژن و تفسیر دادههای اپیژنتیکی استفاده میکنند. همچنین رشد فناوریهای توالییابی نسل جدید و افزایش حجم دادههای ژنومیک، نقش ابزارهای مصورسازی مانند IGV را در استخراج اطلاعات ارزشمند و تصمیمگیریهای پژوهشی پررنگتر کرده است.
هر پژوهشگر فعال در حوزه علوم زیستی، پزشکی و بیوانفورماتیک با یادگیری IGV میتواند دادههای ژنومیک را دقیقتر تفسیر کند، نتایج حاصل از پایپلاینهای تحلیلی را بهتر ارزیابی کند و درک عمیقتری از الگوهای زیستی به دست آورد.
IGV (Integrative Genomics Viewer) یک مرورگر ژنومی رایگان و متنباز است که برای مشاهده و تحلیل دادههای NGS، واریانتها، همترازیها و دادههای اپیژنتیکی استفاده میشود.
بله، IGV بهصورت کاملاً رایگان و متنباز توسط Broad Institute توسعه یافته و برای استفاده پژوهشی و آموزشی در دسترس است.
خیر، نسخه دسکتاپ IGV بهصورت آفلاین کار میکند و میتواند فایلهای محلی را بدون نیاز به اینترنت بارگذاری و تحلیل کند.
IGV برای بررسی سریع فایلهای NGS و اعتبارسنجی واریانتها روی سیستم محلی مناسبتر است، در حالی که UCSC برای دسترسی به دادهها و حاشیهنویسیهای عمومی ژنوم کاربرد بیشتری دارد.
IGV برای مصورسازی دادهها استفاده میشود، در حالی که igvtools وظیفه پیشپردازش، ایندکسگذاری، محاسبه پوشش و بهینهسازی فایلها را بر عهده دارد.
بله، IGV امکان مشاهده پوشش ژنها، بررسی الگوهای Alternative Splicing و تحلیل دادههای RNA-seq را فراهم میکند.

تیم تولید محتوای گروه بیوانفورماتیک وانیار در تلاش است تا بهترین آموزشهای کوتاه در زمینه بیوانفورماتیک و زیستشناسی را تهیه نماید. صحت محتوای این صفحه توسط کارشناسان گروه بیوانفورماتیک وانیار بررسی شده است.




عضویت در مجله وانیار
چطور از جدیدترین آموزشها باخبر شوم؟
با عضویت در مجله بیوانفورماتیک وانیار، برترین آموزشهای بیوانفورماتیک را در لحظه انتشار دریافت کنید.
سلام، وقت بخیر.
چطور میتونیم بهتون کمک کنیم؟
تیم ما آماده پاسخگویی به سوالات شماست.
پشتیبانی 24 ساعته در 7 روز هفته.