ابزار BWA (مخفف Burrows-Wheeler Aligner) بهعنوان یکی از حیاتیترین ابزارها برای فرایند همردیفی (Alignment) خوانشهای کوتاه در آنالیز دادههای توالییابی نسل جدید (NGS) شناخته میشود. با پیشرفت چشمگیر تکنولوژیهای NGS و کاربرد گسترده آنها در پژوهشها و تشخیصهای بالینی، حجم عظیمی از دادههای ژنومی تولید میشود که تحلیل دقیق آنها، نیازمند ابزارهای محاسباتی قدرتمند و کارآمدی مانند این پکیج نرمافزاری است.
BWA که توسط Heng Li در سال ۲۰۰۹ معرفی شد، به یکی از استانداردهای طلایی در حوزه توالییابی تبدیل شده است. این نرمافزار با بهرهگیری از منطق ریاضیاتی و الگوریتم Burrows-Wheeler Transform (BWT)، امکان همردیفی بسیار سریع و دقیق میلیونها خوانش کوتاه را با ژنومهای مرجع بزرگ (مانند ژنوم انسان) فراهم میکند.
بسته BWA به طور کلی شامل ۳ الگوریتم مجزا است: BWA-backtrack (طراحیشده برای خوانشهای ایلومینا تا ۱۰۰ جفتباز)، BWA-SW (برای خوانشهای بلندتر تا یک میلیون جفتباز) و در نهایت BWA-MEM که پیشرفتهترین عضو این مجموعه است. در سالهای اخیر نیز نسخه مستقل و بهینهسازیشده BWA-MEM2 توسعه پیدا کرده که با بازنویسی ساختار کدها، همان دقت الگوریتم MEM را با سرعت پردازشی بسیار بالاتری به محققان ارائه میدهد.
تصور کنید ژنوم انسان یک کتاب دایرةالمعارف بسیار قطور با میلیاردها کلمه است. دستگاههای توالییابی، این کتاب را به میلیونها جمله بسیار خرد و کوچک (به نام خوانش یا Short read) تبدیل میکنند. کار اصلی الگوریتم BWA این است که با سرعتی بینظیر بگردد و پیدا کند که هر کدام از این جملههای خردشده، دقیقاً متعلق به کجای آن دایرةالمعارف بزرگ هستند و این کار را حتی اگر کلماتی جا افتاده باشند (Gap) یا غلط املایی داشته باشند (Mismatch) انجام میدهد.
در روشهای قدیمی، کامپیوتر کل کتاب را کلمه به کلمه میخواند که به حافظه و زمان بسیار زیادی نیاز داشت. اما BWA از یک ترفند ریاضی به نام BWT استفاده میکند.
دادههای ژنتیکی همیشه بینقص نیستند؛ گاهی در آنها جهش رخ داده یا دستگاه توالییاب اشتباه کرده است. برای اینکه BWA بتواند قطعاتی را پیدا کند که ۱۰۰٪ شبیه به ژنوم رفرنس نیستند، از روشی به نام عقبگرد (Backtrack) استفاده میکند:
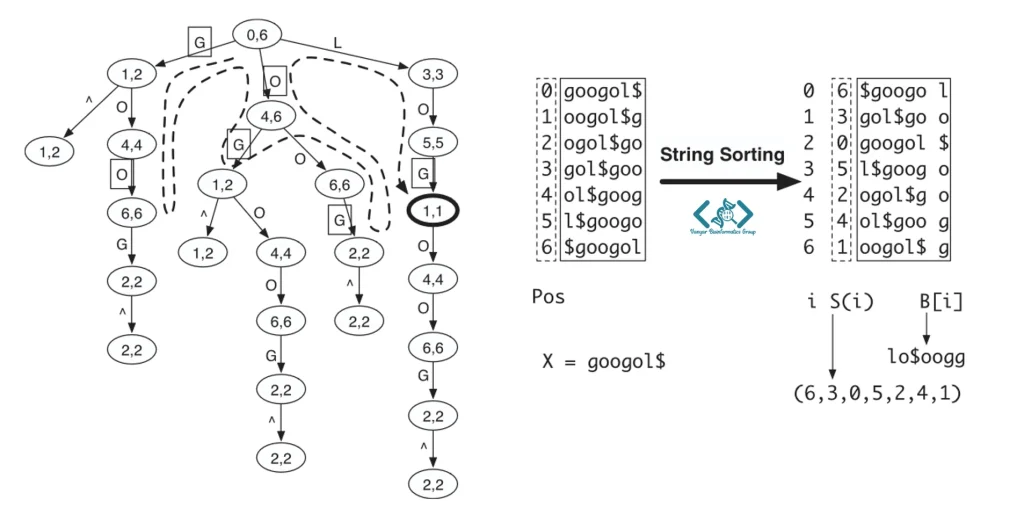
الگوریتم کلمه «GOOGOL» را در قالب یک Prefix trie ذخیره میکند و برای یافتن «LOL» مسیرهای مختلف را با اجازه حداکثر یک عدمتطابق بررسی میکند. در نهایت، با استفاده از Backtracking، کلمه «GOL» را بهعنوان نزدیکترین تطابق پیدا میکند؛ زیرا فقط در یک حرف با «LOL» تفاوت دارد.
در BWT، تمام چرخشهای ممکن یک رشته ساخته و سپس بهترتیب الفبایی مرتب میشوند. با کنار هم قرار دادن آخرین حرف هر ردیف، رشته BWT تولید میشود. این تبدیل حروف مشابه را در کنار هم قرار میدهد و باعث افزایش سرعت جستجو و کاهش مصرف حافظه میشود.
پیش از شروع کار با BWA، لازم است این نرمافزار را روی سیستم خود نصب و پیکربندی کنید. در این بخش، روشهای متداول نصب و اجرای BWA معرفی شدهاند.
BWA تحت مجوز متنباز GPLv3 منتشر شده است. سورسکد BWA را میتوانید از لینک زیر دانلود کنید:
رمز فایل فشرده: www.vanyarbioinf.ir
نصب BWA بسیار ساده است و روی سیستمعاملهای مبتنی بر Unix مانند Linux و macOS بهصورت مستقیم قابل انجام است. کاربران ویندوز نیز میتوانند با استفاده از WSL (Windows Subsystem for Linux) یک محیط لینوکسی ایجاد کرده و BWA را نصب و اجرا کنند.
پس از دانلود فایل فشردهٔ کد منبع BWA، مراحل زیر برای استخراج، کامپایل و بررسی صحت نصب انجام میشود.
tar -xvjf bwa-0.7.17.tar.bz2یا
tar -xf bwa-0.7.17.tar.bz2این دستور فایل فشردهٔ bwa-0.7.17.tar.bz2 را استخراج میکند و پوشهای با نام bwa-0.7.17 شامل کدهای منبع برنامه ایجاد میشود.
cd bwa-0.7.17دستور cd برای تغییر مسیر کاری جاری به پوشهٔ حاوی کد منبع BWA استفاده میشود. تمامی مراحل کامپایل باید در این پوشه انجام شوند.
makeدستور make با استفاده از فایل Makefile موجود در پوشهٔ برنامه، کدهای منبع نوشتهشده به زبان C را کامپایل کرده و فایل اجرایی BWA را تولید میکند. در این مرحله وجود ابزارهای توسعهای نظیر GCC و کتابخانهٔ zlib الزامی است.
پس از پایان موفقیتآمیز فرایند کامپایل، فایل اجرایی bwa در همان پوشه ایجاد خواهد شد.
./bwaپیشوند ./ نشان میدهد که فایل اجرایی موجود در پوشهٔ فعلی اجرا شود. در صورت نصب صحیح، اطلاعاتی مشابه نمونهٔ زیر نمایش داده خواهد شد:
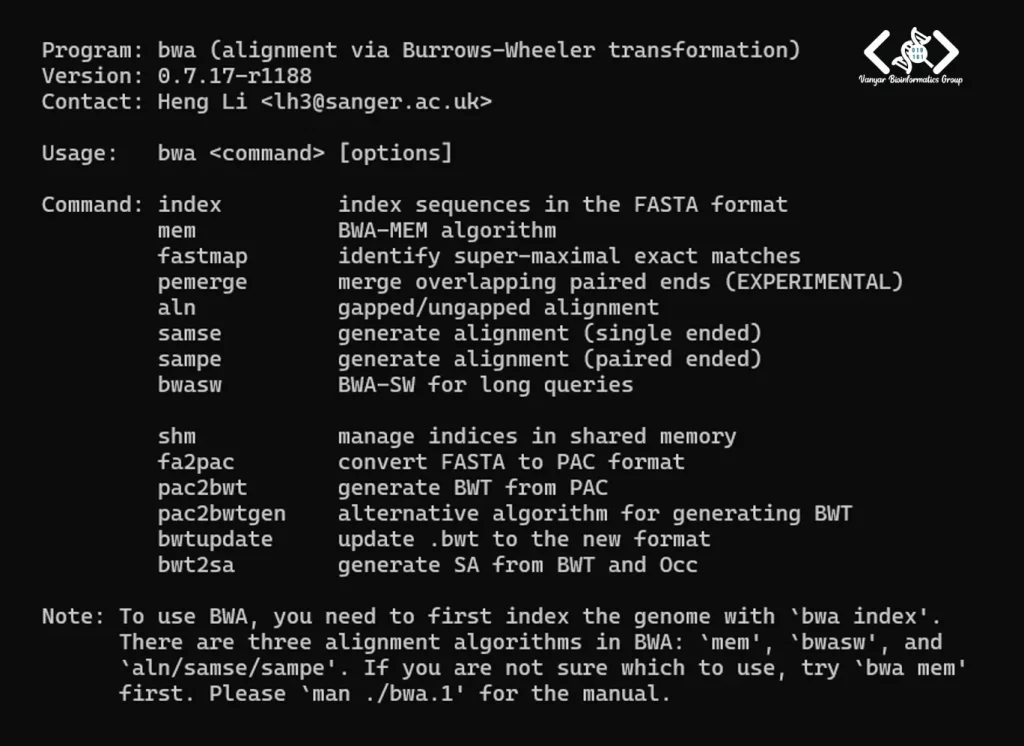
نمایش نام برنامه، شماره نسخه و راهنمای استفاده نشاندهندهٔ موفقیتآمیز بودن فرایند کامپایل و آمادهبودن BWA برای استفاده در تحلیل دادههای توالییابی است.
یکی از سادهترین روشهای نصب BWA، استفاده از Bioconda است. Bioconda یک مخزن تخصصی برای نرمافزارهای بیوانفورماتیکی است که امکان نصب آسان برنامهها و وابستگیهای آنها را فراهم میکند. برای نصب BWA کافی است دستور زیر اجرا شود:
conda install -c bioconda bwaاین دستور، مشخص میکند که بسته از کانال Bioconda دریافت شود و Conda بهصورت خودکار نرمافزار و وابستگیهای موردنیاز آن را نصب میکند. این روش نسبت به کامپایل از کد منبع سادهتر بوده و بهطور گسترده در محیطهای بیوانفورماتیکی مورد استفاده قرار میگیرد.
پس از اتمام نصب، میتوان با اجرای دستور زیر از صحت نصب اطمینان حاصل کرد:
bwaدر صورت نصب موفق، اطلاعاتی شامل نسخه برنامه و راهنمای استفاده از دستورات BWA نمایش داده خواهد شد.
الگوریتم BWA-MEM که در سال ۲۰۱۳ معرفی شد، نسخه پیشرفته و بهبودیافتهای از مجموعه BWA است که بر پایه یافتن بیشترین تطابقهای دقیق (Maximal Exact Matches یا MEMs) عمل میکند. در حالی که نسخههای قبلی مانند BWA-backtrack محدود به خوانشهای کوتاه (زیر ۱۰۰ جفتباز) بودند، الگوریتم BWA-MEM به عنوان یک استاندارد طلایی برای همترازی خوانشهای ۷۰ جفتباز تا چندین مگاباز طراحی و بهینه شده است.
عملکرد BWA-MEM نه تنها بسیار سریعتر از نسخههای پیشین است، بلکه دقت بسیار بالاتری در مدیریت مناطق چالشبرانگیزِ ژنوم (مانند مناطقی با حذف و اضافههای متعدد (Indels) یا ناهمخوانیهای زیاد) دارد. این الگوریتم به جای تنظیمات دستی پیچیده، از یک سیستم امتیازدهی منعطف بهره میبرد که به خوبی با کیفیتها و طولهای مختلف خوانشها سازگار میشود.
از ویژگیهای برجسته BWA-MEM، توانایی شناسایی خوانشهای کایمریک (Chimeras) و دوپاره (Split-reads) است که آن را به ابزاری حیاتی در مطالعات تنوع ساختاری ژنوم (Structural Variants) تبدیل میکند. این الگوریتم به جای جستجوی کورکورانه در کل ژنوم، از رویکرد بذرپاشی و گسترش (Seed-and-Extend) استفاده میکند؛ به این معنا که ابتدا مناطق امیدبخش (بذرها) را شناسایی کرده و سپس با استفاده از الگوریتم محلی Smith-Waterman، تطبیق دقیق را فقط در همان نواحی انجام میدهد.
BWA-MEM2 نسل جدید الگوریتم BWA-MEM است که با هدف افزایش سرعت الاینمنت توالیها توسعه یافته است. این ابزار از نظر منطق همترازی و نتایج خروجی با BWA-MEM یکسان بوده و فایلهای SAM تولیدشده توسط آن معادل خروجی BWA-MEM هستند. با این حال، بهینهسازیهای انجامشده در ساختار کد و بهرهگیری بهتر از قابلیتهای پردازندههای مدرن باعث شده است که BWA-MEM2 بسته به نوع دیتا و سختافزار مورد استفاده، بین 1.3 تا 3.1 برابر سریعتر عمل کند.
پژوهشگران آزمایشگاه محاسبات موازی Intel، ابزار BWA-MEM2 را توسعه داده و آن را تحت مجوز متنباز MIT منتشر کردهاند. امروزه این ابزار در بسیاری از پایپلاینهای تحلیل دادههای NGS بهعنوان جایگزینی سریعتر برای BWA-MEM مورد استفاده قرار میگیرد.
هر یک از سه الگوریتم اصلی BWA (شامل BWA-backtrack ، BWA-SW و BWA-MEM) برای طول و نوع خاصی از دادههای توالییابی مناسب هستند. در تمامی این الگوریتمها، نخست باید ژنوم مرجع ایندکس شود و سپس فرآیند همترازی انجام گیرد.
پیش از شروع همترازی، BWA باید ساختار FM-index را برای ژنوم رفرنس ایجاد کند:
bwa index ref.fa
این دستور فایل رفرنس در قالب FASTA را پردازش کرده و فایلهای ایندکس موردنیاز برای جستجو و الاینمنت را تولید میکند.
این الگوریتم در حال حاضر رایجترین روش استفاده از BWA برای دادههای Illumina محسوب میشود.
bwa mem ref.fa reads.fq > aln.sam
bwa mem ref.fa read1.fq read2.fq > aln-pe.sam
در این دستورات، فایلهای FASTQ به ژنوم مرجع نگاشت شده و نتیجه در قالب SAM ذخیره میشود.
برای دادههای قدیمی Illumina با طول کمتر از حدود 70 جفتباز میتوان از الگوریتم BWA-backtrack استفاده کرد.
bwa aln ref.fa reads.fq > reads.sai
bwa samse ref.fa reads.sai reads.fq > aln-se.sam
bwa aln ref.fa read1.fq > read1.sai
bwa aln ref.fa read2.fq > read2.sai
bwa sampe ref.fa read1.sai read2.sai read1.fq read2.fq > aln-pe.sam
امروزه با افزایش طول خوانشهای Illumina، محققان نسبت به گذشته کمتر از BWA-backtrack استفاده میکنند.
توسعهدهندگان، الگوریتم BWA-SW را برای همترازی توالیهای طولانی طراحی کردهاند:
bwa bwasw ref.fa long_read.fq > aln.sam
با این حال، در اکثر کاربردهای امروزی، متخصصان BWA-MEM را جایگزین BWA-SW کردهاند و کمتر آن را به کار میبرند.
نسخههای اولیه BWA-MEM از دادههای حاصل از فناوریهای PacBio و Oxford Nanopore نیز پشتیبانی میکردند:
با این حال، توسعهدهندگان BWA اکنون استفاده از Minimap2 را برای الاینمنت دادههای PacBio و Nanopore توصیه میکنند. Minimap2 علاوه بر پشتیبانی از قابلیتهای اصلی BWA-MEM، سرعت بسیار بالاتری داشته و دقت بیشتری در همترازی خوانشهای بلند ارائه میدهد. بنابراین، برای پروژههای مبتنی بر توالییابی Long-read، ابزار Minimap2 معمولاً انتخاب اول محسوب میشود.
همانند BWA، پیش از انجام همترازی باید ژنوم مرجع ایندکس شود اما توجه کنید که: فرایند ایندکسسازی به حافظه نسبتاً زیادی نیاز دارد و تقریباً به 28 برابر اندازه فایل مرجع حافظه RAM احتیاج دارد. به همین دلیل برای ژنومهای بزرگ مانند ژنوم انسان، استفاده از سیستمهای دارای حافظه کافی ضروری است!
ساختار اجرای BWA-MEM2 تقریباً مشابه BWA-MEM است.
bwa-mem2 mem reference.fa reads.fastq > output.sambwa-mem2 mem reference.fa read1.fastq read2.fastq > output.samیکی از مهمترین مزایای BWA-MEM2، بهرهگیری مؤثر از پردازش موازی است. تعداد هستههای مورد استفاده با گزینه -t مشخص میشود:
bwa-mem2 mem -t 16 reference.fa read1.fastq.gz read2.fastq.gz > output.samدر این مثال، 16 هسته پردازشی برای انجام همترازی استفاده میشود که میتواند زمان اجرای تحلیل را بهطور قابل توجهی کاهش دهد.
| نوع دیتا | الگوریتم پیشنهادی |
|---|---|
| Illumina کوتاه (<70 bp) | BWA-backtrack |
| Illumina معمولی (≥70 bp) | BWA-MEM یا BWA-MEM2 |
| Oxford Nanopore | PacBio CLR | PacBio HiFi | Minimap2 |
| Assembly Contigs (کانتیگهای حاصل از اسمبلی) | BWA-MEM یا Minimap2 |
در حال حاضر، BWA-MEM2 برای دادههای Illumina و Minimap2 برای دادههای Long-read رایجترین انتخابها در پایپلاینهای مدرن بیوانفورماتیکی هستند.
پیش از مقایسه این ابزارها، باید توجه داشت که اصطلاح BWA در متون مختلف ممکن است به دو معنا به کار رود: گاهی به کل نرمافزار Burrows-Wheeler Aligner اشاره دارد و گاهی منظور الگوریتم اولیه آن (که نخستین بار در سال 2009 معرفی شد) است. برای جلوگیری از ابهام، در این بخش از نام BWA-backtrack برای اشاره به الگوریتم اولیه BWA استفاده میشود.
| BWA-MEM2 | BWA-MEM | BWA-backtrack | |
|---|---|---|---|
| دستورات اصلی | mem | mem | aln ، samse ، sampe |
| طول خوانش مناسب | بیش از 70 bp | بیش از 70 bp | تا حدود 100 جفتباز |
| دقت الاینمنت | مشابه BWA-MEM | بالاتر از BWA-backtrack | مناسب برای خوانشهای کوتاه |
| سرعت اجرا | حدود 1.3 تا 3.1 برابر سریعتر از BWA-MEM | بیشتر از BWA-backtrack | کمترین |
| کاربرد امروزی | جایگزین بهینهشده BWA-MEM | بسیار رایج | محدود |
BWA-MEM برای خوانشهای Illumina با طول 70 تا 100 جفتباز و بیشتر، عملکرد بهتری نسبت به BWA-backtrack ارائه میدهد و بهعنوان الگوریتم پیشفرض BWA شناخته میشود. BWA-MEM2 نیز همان نتایج همترازی BWA-MEM را تولید میکند، اما با بهرهگیری از بهینهسازیهای معماری پردازنده، سرعت اجرای بالاتری دارد.
پژوهشگران و متخصصان بیوانفورماتیک طی بیش از یک دهه گذشته از BWA بهعنوان یکی از استانداردهای اصلی الاینمنت توالیها استفاده کردهاند و همچنان آن را در بسیاری از پایپلاینهای تحلیل دادههای NGS به کار میبرند. اگرچه الگوریتم اولیه BWA-backtrack نقش مهمی در توسعه روشهای همترازی مدرن داشته است، امروزه BWA-MEM و بهویژه BWA-MEM2 به دلیل سرعت بالاتر، دقت بیشتر و پشتیبانی بهتر از خوانشهای طولانیتر، انتخابهای مناسبتری برای دادههای Illumina محسوب میشوند.
انتخاب صحیح الگوریتم بر اساس نوع دیتا، طول خوانشها و منابع محاسباتی در دسترس، تأثیر مستقیمی بر کیفیت نتایج تحلیلهای ژنومی خواهد داشت و میتواند دقت مراحل بعدی مانند فراخوانی واریانتها (variant calling) و تحلیلهای ساختاری را بهبود بخشد.
BWA یا Burrows-Wheeler Aligner یکی از پرکاربردترین ابزارهای الاینمنت توالی است که برای نگاشت خوانشهای حاصل از توالییابی به ژنوم رفرنس استفاده میشود. این ابزار در بسیاری از پایپلاینهای تحلیل NGS به کار میرود.
خروجی و دقت هر دو الگوریتم کاملاً یکسان است؛ اما BWA-MEM2 با استفاده بهینه از پردازندههای مدرن، فرایند الاینمنت را بین ۱.۳ تا ۳.۱ برابر سریعتر انجام میدهد.
الگوریتم BWA-MEM پرکاربردترین عضو پکیج BWA برای الاینمنت خوانشهای بالای ۷۰ جفتباز با ژنوم رفرنس است که کاربرد وسیعی در پایپلاینهای شناسایی واریانتها (مانند توالییابی اگزوم) دارد.
این ابزار نسخه بازنویسیشده و فوقسریع BWA-MEM است که با بهرهگیری از معماری پردازندههای مدرن، همان خروجی و دقت الاینمنت را با سرعتی تا ۳.۱ برابر بیشتر در پروژههای بزرگ ژنومیکس پردازش میکند.
برای خوانشهای باکیفیت و استاندارد ایلومینا (طول بالای ۷۰ جفتباز)، الگوریتمهای پیشرفته BWA-MEM و BWA-MEM2 استاندارد طلایی و بهترین انتخاب هستند.
توسعهدهندگان نرمافزار توصیه میکنند برای دادههای Long-read (مانند Nanopore یا PacBio) به جای BWA، از نرمافزار تخصصی و سریعتر Minimap2 استفاده شود.
بله، اما معمولاً توصیه نمیشود. BWA برای الاینمنت DNA طراحی شده و splice-aware نیست؛ بنابراین خوانشهای عبوری از مرز اگزونها را بهخوبی مدیریت نمیکند. برای دادههای RNA-seq معمولاً از ابزارهایی مانند HISAT2 یا STAR استفاده میشود.

تیم تولید محتوای گروه بیوانفورماتیک وانیار در تلاش است تا بهترین آموزشهای کوتاه در زمینه بیوانفورماتیک و زیستشناسی را تهیه نماید. صحت محتوای این صفحه توسط کارشناسان گروه بیوانفورماتیک وانیار بررسی شده است.




عضویت در مجله وانیار
چطور از جدیدترین آموزشها باخبر شوم؟
با عضویت در مجله بیوانفورماتیک وانیار، برترین آموزشهای بیوانفورماتیک را در لحظه انتشار دریافت کنید.
سلام، وقت بخیر.
چطور میتونیم بهتون کمک کنیم؟
تیم ما آماده پاسخگویی به سوالات شماست.
پشتیبانی 24 ساعته در 7 روز هفته.