HISAT2 (Hierarchical Indexing for Spliced Alignment of Transcripts) یکی از ابزارهای سریع، حساس و کارآمد بیوانفورماتیک برای الاینمنت خوانشهای توالییابی نسل جدید با ژنوم انسان است. این نرمافزار که توسط Daehwan Kim و همکارانش توسعه یافته، برای نگاشت (mapping) دیتای توالییابی whole-genome ، transcriptome و exome طراحی شده و علاوه بر ژنوم رفرنس خطی، میتواند تنوع ژنتیکی موجود در جمعیت انسانی را نیز در فرایند همترازی لحاظ کند.
نوآوری اصلی HISAT2 در بهرهگیری از یک چارچوب گرافمحور برای ایندکسگذاری ژنوم است؛ چارچوبی که با استفاده از Graph FM Index (GFM) و نسخهی سلسلهمراتبی آن یعنی Hierarchical Graph FM Index (HGFM)، امکان جستوجوی سریع را در میان ژنوم رفرنس و تعداد زیادی از واریانتهای ژنتیکی فراهم میکند. این ویژگی باعث میشود HISAT2 در مقایسه با روشهای متکی بر یک رفرنس خطی، بهویژه در نواحی بسیار چندشکل و پرتنوع ژنومی، از حساسیت و دقت بالاتری برخوردار باشد.
HISAT2 با ترکیب یک ایندکس سراسری و مجموعهای از ایندکسهای محلی کوچک، قادر است خوانشها (reads) را بهصورت مؤثر و دقیق همتراز کند. همچنین خروجی آن در قالب SAM ارائه میشود که سازگاری آن را با ابزارهای متداولی مانند SAMtools و GATK تضمین میکند. در مجموع، HISAT2 بهعنوان ابزاری متنباز و عملی، یکی از روشهای مهم و پیشرفته برای همترازی دادههای توالییابی و تحلیل واریانتهای ژنتیکی در مقیاس ژنوم بهشمار میرود.
در این مقاله، به بررسی جامع HISAT2 از مبانی الگوریتمی و معماری گرافمحور آن تا کاربردهای عملی در آنالیز دادهها خواهیم پرداخت. همچنین نحوه دانلود، ساخت ایندکس، اجرای دستورات خط فرمان و تفسیر نتایج الاینمنت را آموزش میدهیم. این راهنما برای دانشجویان، پژوهشگران و متخصصان بیوانفورماتیک که با دادههای ژنومیک، ترنسکریپتومیک (RNA-Seq) و تحلیل واریانتها سروکار دارند، منبعی جامع و ارزشمند خواهد بود.
در این بخش، الگوریتمهای زیربنایی HISAT2 را شرح میدهیم:
این ابزار از یک ساختار داده جدید مبتنی بر گراف (graph-based data structure) و یک الگوریتم الاینمنت استفاده میکند تا خوانشهای توالییابی را به سرعت و با دقت بالا با ژنوم و مجموعه بزرگی از واریانتهای کوچک تطبیق دهد. همچنین، HISAT2 یک الگوریتم ایندکسگذاری نوین برای توالیهای تکراری در ژنوم معرفی کرده است. این الگوریتم به گونهای عمل میکند که الاینمنت یک خوانش تکراری ابتدا به یک مکان واحد نگاشت میشوند و سپس به طور کامل بازیابی میگردند.
ژنوم رفرنس انسان (مثل GRCh38) تنها بر اساس اطلاعات ژنتیکی چند نفر ساخته شده است و تنوع ژنتیکی کل جمعیت را شامل نمیشود. این موضوع باعث میشود ابزارهای اجرای الاینمنت در مواجهه با ژنومهای جدید و متفاوت دچار خطا شوند.
الگوریتم HISAT2 بهجای استفاده از یک رشته خطی ساده، از ساختار گراف (Graph) استفاده میکند. در این گراف، تمام جهشها، حذفها و اضافهشدنهای ژنتیکی (Variants) بهعنوان مسیرهای جایگزین و فرعی تعریف میشوند.
برای اینکه جستجو در این گرافِ پیچیده سریع و بهینه باشد، دو کار مهم انجام میشود:
با این معماری، HISAT2 موفق شده کل ژنوم انسان بهعلاوه ۱۴.۵ میلیون تنوع ژنتیکی شناختهشده را تنها در ۶.۲ گیگابایت حافظه جا دهد. این روش نسبت به ابزارهای قبلی دقت بسیار بالاتری دارد و با وجود پردازش اطلاعاتِ بهمراتب بیشتر، افت سرعت چندانی ندارد.
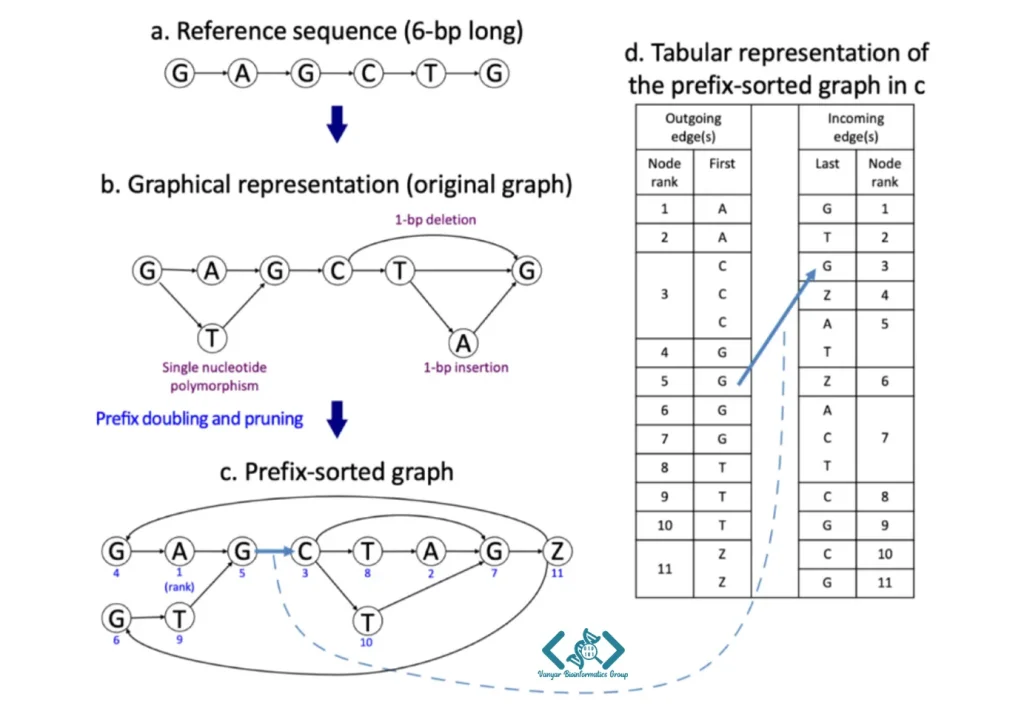
در تصویر بالا، تبدیل یک توالی ژنتیکی ساده به یک ساختار داده فشرده و هوشمند را مشاهده میکنید که مراحل آن عبارتند از:
این سادهترین حالت ممکن است. یک توالی ۶ حرفی (G-A-G-C-T-G) را میبینید که بهصورت یک خط مستقیم و بدون هیچگونه تنوع یا جهش ژنتیکی رسم شده است.
حالا برای اینکه تفاوتهای ژنتیکی بین انسانهای مختلف پوشش داده شود، ۳ نوع تغییر ژنتیکی (Variant) به عنوان مسیرهای جایگزین و فرعی به توالی اضافه شدهاند:
گراف مرحله قبل برای کامپیوتر کمی گیجکننده است. برای جستجوی سریعتر، ساختار گراف بر اساس ترتیب الفبایی مرتب میشود. به هر دایره (گره) یک شماره جدید (اعداد آبی رنگ زیر دوایر) داده میشود. این رتبهبندی کمک میکند تا جستجو با نظم مشخصی انجام شود. حرف Z هم در انتها اضافه شده تا به سیستم بگوید توالی دقیقاً در اینجا به پایان میرسد.
در این مرحله، کل گراف بدون از دست رفتن اطلاعات به یک جدول فشرده تبدیل میشود تا حافظه بسیار کمی اشغال کند:
در ژنوم انسان توالیهای تکراری زیادی وجود دارد. وقتی قطعات خوانششده به این بخشها میرسند، ممکن است با دهها یا صدها نقطه مختلف در ژنوم تطابق داشته باشند (حدود ۱ تا ۳ درصد از دادهها این حالت را دارند). ابزارهای معمولی (مثل BWA) یا فقط یکی از این نقاط را بهصورت تصادفی گزارش میدهند، یا اگر بخواهند همه را گزارش دهند، حجم فایل خروجی به شکل غیرقابلکنترلی بالا میرود.
HISAT2 تمام توالیهای دقیقاً مشابه که در جاهای مختلف ژنوم پراکنده شدهاند را با هم ترکیب میکند و آنها را بهعنوان یک توالی نماینده در نظر میگیرد. حالا اگر یک Read متعلق به این بخشهای تکراری باشد، بهجای اینکه مثلاً به ۱۰۰ مکان مختلف الاین شود، فقط یکبار به این توالی نماینده متصل میشود.
این ترفند باعث میشود فایلهای خروجی بهشدت سبک شوند.
نرمافزار HISAT2 کتابخانهها و رابطهای برنامهنویسی اختصاصی برای زبانهای پایتون، جاوا و C++ ارائه میدهد تا در مراحل بعدی آنالیز ژنومیک (مثل شناسایی واریانتها یا Variant Calling)، سیستم بتواند در صورت نیاز، مکانهای واقعی این قطعات را روی ژنوم اصلی با سرعت بالا بازیابی کند.
نسخههای باینری را میتوانید از لینکهای زیر دانلود کنید. نسخههای باینری برای معماریهای اینتل (x86_64) در سیستمعاملهای لینوکس و مک (Mac OS X) ارائه شدهاند.
رمز فایل فشرده: www.vanyarbioinf.ir
ابزار HISAT2 در سیستمعاملهای لینوکس، مک OS و ویندوز (از طریق WSL) قابل اجراست.
توجه: یک سیستم با حداقل 8 گیگابایت RAM برای ژنومهای کوچک تا متوسط و 16 گیگابایت یا بیشتر برای ژنومهای بزرگ مانند انسان توصیه میشود. همچنین، داشتن چندین هسته پردازشی میتواند سرعت آنالیز را به طور قابل توجهی افزایش دهد.
فرایند پیش از الاینمنت شامل دو گام اساسی است: دسترسی به ژنوم رفرنس و پیکربندی ایندکسهای اختصاصی. در ادامه، نقشهی راهِ اجرایی این دو مرحله را بررسی میکنیم.
اولین قدم در استفاده از HISAT2، تهیه ژنوم رفرنس مناسب است. میتوانید ژنوم مرجع را از دیتابیسهای معتبر مانند ENSEMBL ، UCSC یا NCBI دانلود کنید.
دومین قدم قبل از اجرای الاینمنت، ساخت ایندکس است. این مرحله به HISAT2 اجازه میدهد تا جستجوی سریع و کارامدی را در ژنوم رفرنس انجام دهد. ساخت ایندکس فقط یک بار برای هر ژنوم رفرنس انجام میشود.
HISAT2 بر اساس اندازه ژنوم رفرنس، تصمیم میگیرد از چه نوع ایندکسی استفاده کند:
.ht2 ذخیره میشوند و از اعداد ۳۲ بیتی استفاده میکنند..ht2l ذخیره میشوند و از اعداد ۶۴ بیتی استفاده میکنند.*** کاربر نیازی به دخالت ندارد؛ اسکریپتهای HISAT2 به طور خودکار نوع ایندکس مناسب را تشخیص میدهند و آن را میسازند.
ساختار کلی دستور به این صورت است:
hisat2-build [options]* <reference_in> <ht2_base>
<reference_in>: لیست فایلهای FASTA (جدا شده با کاما) یا خودِ توالیها (اگر از گزینه -c استفاده شود).<ht2_base>: نام پایه (Basename) که میخواهید فایلهای خروجی با آن نامگذاری شوند.توجه: برای کاهش مصرف حافظه و سادهتر شدن فرایند کار، امکان دانلود ایندکسهای آماده بهصورت رایگان نیز وجود دارد؛ بنابراین لازم نیست حتماً ایندکس را خودتان بسازید 🙂
اکنون میتوانیم به محیط خط فرمان وارد شویم … در بخش پیش رو، میآموزیم که چگونه با تعیین فایلهای ورودی و بهکارگیری گزینههای کنترلی، فرایند الاینمنت را متناسب با نیاز خود تنظیم کنیم.
قالب کلی دستور عبارتست از:
hisat2 [options]* -x <hisat2-idx> {-1 <m1> -2 <m2> | -U <r> | --sra-acc <SRA accession number>} [-S <hit>]
یعنی HISAT2 برای اجرا به این موارد نیاز دارد (آرگومانهای اصلی):
-x)-1 و -2-U--sra-acc (این گزینه برای زمانی است که میخواهید دادهها را مستقیم از SRA بگیرید)-S (اگر این آپشن را تعیین نکنید، خروجی به صورت پیشفرض به stdout میرود؛ یعنی در ترمینال چاپ میشود)از آپشنهای این دستور میتوان به سه مورد زیر در رابطه با تعیین فرمت فایل ورودی اشاره کرد:
-q (پیشفرض): یعنی فایلهای شما FASTQ هستند (معمولاً دارای کیفیت توالی یا Quality Score).-f: یعنی فایلهای شما FASTA هستند. چون FASTA اطلاعات کیفیت ندارد، HISAT2 خودش به صورت خودکار --ignore-quals را فعال میکند.-c: بسیار جالب است؛ اگر میخواهید توالیها را مستقیماً در ترمینال تایپ کنید (مثلاً برای تست سریع)، از این گزینه استفاده میکنید.فرض کنید میخواهید یک فایل FASTQ را تراز کنید، ۵ بازِ اول آن را حذف کنید و فقط ۵۰۰۰ خوانش اول را پردازش کنید:
hisat2 -x genome_index -q -1 reads_1.fq -2 reads_2.fq -5 5 -u 5000 -S output.sam--trim5 یا -5 یک گزینه در HISAT2 است که به شما اجازه میدهد تعداد مشخصی از بازها (bases) را از انتهای چپ (سمت ۵’) هر خوانش حذف کنید، قبل از اینکه HISAT2 شروع به تراز کردن کند.
گزینه -u (یا --upto) در HISAT2 به شما اجازه میدهد تعداد محدودی از خوانشها را پردازش کنید و سپس متوقف شوید.
در پایان همترازسازی، خلاصهای از نتایج برای شما چاپ میشود. تفسیر آن به این صورت است:
20000 reads; of these:
20000 (100.00%) were unpaired; of these:
1247 (6.24%) aligned 0 times
18739 (93.69%) aligned exactly 1 time
14 (0.07%) aligned >1 times
93.77% overall alignment rate
نتایج نشان میدهد: که 6.24% اصلاً مپ نشدهاند | 93.69% دقیقاً یک بار مپ شدهاند | 0.07% به بیش از یک محل مپ شدهاند.
بنابراین:
93.77% overall alignment rate
یعنی حدود 93.8٪ از خوانشها حداقل یک بار روی ژنوم رفرنس مپ شدهاند.
10000 reads; of these:
10000 (100.00%) were paired; of these:
650 (6.50%) aligned concordantly 0 times
8823 (88.23%) aligned concordantly exactly 1 time
527 (5.27%) aligned concordantly >1 times
در دادههای paired-end، دو mate باید فاصله مناسبی از هم داشته باشند و جهت (orientation) مورد انتظار را داشته باشند. اگر هر دو شرط برقرار باشد، جفت خوانش concordant محسوب میشود.
از 10000 جفت خوانش، 650 تا concordant map نشده | 8823 تا concordant و یکتا map شده | 527 تا concordant و چندمحلی map شده.
650 pairs aligned concordantly 0 times; of these:
34 (5.23%) aligned discordantly 1 time
از 650 جفتی که concordant مپ نشدهاند، 34 جفت به صورت discordant مپ شدهاند. یعنی هر دو mate مپ شدهاند ولی فاصله یا جهت آنها با انتظار HISAT2 سازگار نیست.
616 pairs aligned 0 times concordantly or discordantly
زیرا:
650 - 34 = 616
این 616 جفت نه concordant و نه discordant مپ نشدهاند.
1232 mates make up the pairs
هر جفت دو mate دارد:
616 × 2 = 1232
سپس HISAT2 هر mate را جداگانه بررسی میکند:
660 (53.57%) aligned 0 times
571 (46.35%) aligned exactly 1 time
1 (0.08%) aligned >1 times
نرخ الاینمنت کلی:
96.70% overall alignment rate
این مقدار درصد خوانشهایی است که در نهایت حداقل یک alignment معتبر پیدا کردهاند (چه به صورت concordant، چه discordant، چه به صورت mate منفرد). بنابراین این عدد صرفاً برابر با درصد concordant alignment نیست.
HISAT-3N نسخهای توسعهیافته از HISAT2 است که برای همترازسازی دادههای حاصل از فناوریهای Nucleotide Conversion Sequencing (NC-Seq) طراحی شده است. در این فناوریها، طی آمادهسازی نمونه یا فرایندهای بیولوژیکی خاص، برخی نوکلئوتیدها به نوکلئوتیدهای دیگری تبدیل میشوند؛ برای مثال تبدیل سیتوزین به تیمین در Bisulfite Sequencing (BS-seq) یا تبدیل تیمین به سیتوزین در SLAM-seq. این تغییرات باعث میشوند ابزارهای همترازسازی معمولی مانند HISAT2 ، STAR یا BWA این بازهای تبدیلشده را بهعنوان mismatch تفسیر کنند و در نتیجه دقت همترازسازی کاهش یابد. HISAT-3N با استفاده از یک راهبرد «سهنوکلئوتیدی» (3-Nucleotide Alignment) این مشکل را برطرف کرده و امکان همترازسازی دقیق خوانشهای حاصل از انواع فناوریهای مبتنی بر تبدیل نوکلئوتید را فراهم میکند.
برخلاف ابزارهایی مانند Bismark یا SLAM-DUNK که هر کدام برای یک فناوری خاص توسعه یافتهاند، HISAT-3N یک چارچوب عمومی ارائه میدهد که میتواند انواع دادههای NC-Seq شامل BS-seq ، TAPS ، SLAM-seq و سایر روشهای مشابه را برای دادههای DNA و RNA پردازش کند. این ابزار با بهرهگیری از زیرساخت ایندکسگذاری سلسلهمراتبی و الگوریتمهای بهینه HISAT2، سرعت بالا، دقت بیشتر، مقیاسپذیری مناسب و مصرف حافظه کمتری نسبت به بسیاری از ابزارهای موجود ارائه میدهد. به همین دلیل HISAT-3N بهعنوان یکی از پیشرفتهترین گزینهها برای همترازسازی دادههای مبتنی بر تبدیل نوکلئوتید شناخته میشود و میتواند نقش مهمی در مطالعات اپیژنتیک، RNA Editing، متیلاسیون DNA و بررسی فرایندهای سلولی پویا ایفا کند.
از بین ابزارهای امروزی، STAR مهمترین رقیب HISAT2 است و Subread نیز گزینهای معتبر محسوب میشود. TopHat2 بیشتر ارزش تاریخی دارد و امروزه دیگر توصیه نمیشود.
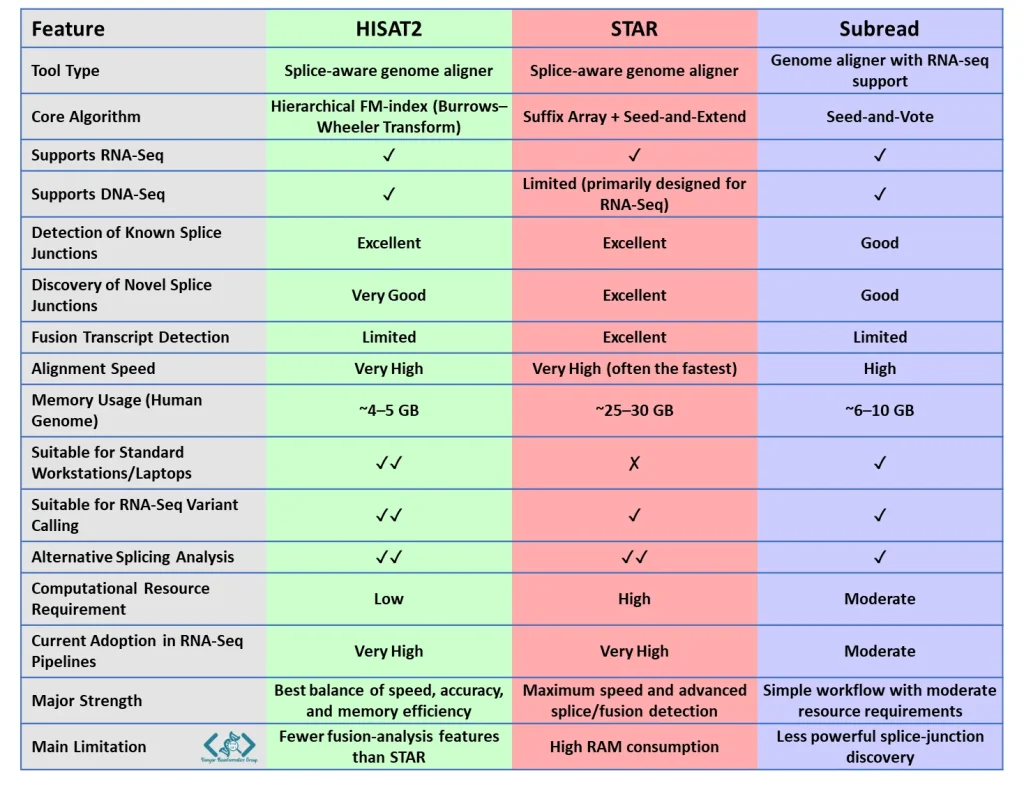
HISAT2 و STAR دو الاینر پرکاربرد RNA-Seq هستند که دقت مشابهی در تشخیص محلهای پیرایش (splice junctions) دارند. STAR معمولاً سریعتر است و قابلیتهای پیشرفتهتری برای شناسایی fusion transcriptها و splice junctionهای جدید ارائه میدهد، اما به حافظه بسیار بیشتری نیاز دارد. در مقابل، HISAT2 با مصرف حافظه حدود 4 تا 5 گیگابایت، دقتی مشابه STAR را حفظ میکند و به همین دلیل یکی از متعادلترین گزینهها برای تحلیل دادههای RNA-Seq محسوب میشود.
HISAT2 یکی از مهمترین همترازکنندههای (Aligner) نسل جدید است که با ترکیب سرعت بالا، دقت مناسب و مصرف حافظه پایین، جایگاه ویژهای در تحلیل دیتای ژنومی و ترنسکریپتومی به دست آورده است. معماری مبتنی بر گراف و استفاده از ایندکسهای سلسلهمراتبی به این ابزار امکان میدهد تا علاوه بر ژنوم رفرنس، تنوع ژنتیکی موجود در جمعیتها را نیز در فرایند الاینمنت در نظر بگیرد. این ویژگی در کنار پشتیبانی از همترازسازی splice-aware، ابزار HISAT2 را به یکی از گزینههای اصلی برای تحلیل RNA-Seq، بررسی رویدادهای پیرایشی و تولید دادههای مناسب برای تحلیلهای پاییندستی تبدیل کرده است.
انتخاب صحیح پارامترهای همترازسازی، استفاده از ژنوم رفرنس مناسب و ارزیابی دقیق کیفیت نتایج، تأثیر مستقیمی بر اعتبار تحلیلهای بعدی خواهد داشت. از این رو، آشنایی با قابلیتهای HISAT2 میتواند به پژوهشگران کمک کند تا از این ابزار بهصورت مؤثر در پروژههای مختلف بیوانفورماتیک و ژنومیک بهره ببرند.
HISAT2 یک نرمافزار سریع و دقیق برای الاینمنت خوانشهای توالییابی نسل جدید است. کاربرد اصلی این ابزار، مپ یا نگاشت کردن دیتای حاصل از توالییابی کل ژنوم (WGS)، اگزوم (WES) و بهویژه ترنسکریپتوم (RNA-Seq) روی ژنوم رفرنس است.
بله، HISAT2 کاملاً رایگان و متنباز است. میتوانید این ابزار را دانلود کنید و بهراحتی روی سیستم خود (لینوکس، مک یا ویندوز از طریق WSL) نصب و اجرا نمایید.
ابزار BWA توالیها را روی یک خط صاف (ژنوم خطی) مپ میکند، اما HISAT2 تنوعهای ژنتیکی جمعیت را به صورت یک گراف (مسیرهای فرعی) در نظر میگیرد تا دقت بالاتر برود. همچنین، HISAT2 نسخه جدیدتر، بسیار سریعتر و دقیقتر TopHat2 است (TopHat2 منسوخ شده).
برای الاینمنت فقط به ۶ تا ۸ گیگابایت رم نیاز دارید (قابل اجرا روی کامپیوترهای خانگی).اما برای ساخت ایندکس گرافمحور جدید، حدود ۲۰۰ گیگابایت رم لازم است (البته میتوانید ایندکسهای آماده را رایگان دانلود کنید تا نیازی به این مقدار رم نباشد).
هر دو بسیار دقیق هستند، اما تفاوت اصلی در مصرف رم است. STAR سرعت فوقالعادهای دارد اما به ۳۰ تا ۴۰ گیگابایت رم نیاز دارد. HISAT2 سرعت بسیار خوب و دقت مشابهی دارد، اما فقط با ۵ گیگابایت رم اجرا میشود. پس اگر محدودیت سختافزاری دارید، HISAT2 بهترین انتخاب است.
HISAT-3N نسخه تخصصی HISAT2 برای تحلیل دادههای تبدیل نوکلئوتیدی (NC-Seq) مثل Bisulfite Sequencing است. HISAT2 معمولی این تغییرات را به عنوان «خطا» رد میکند، اما HISAT-3N با الگوریتم خاص خود، این تغییرات را بهدرستی تشخیص میدهد و الاین میکند.

تیم تولید محتوای گروه بیوانفورماتیک وانیار در تلاش است تا بهترین آموزشهای کوتاه در زمینه بیوانفورماتیک و زیستشناسی را تهیه نماید. صحت محتوای این صفحه توسط کارشناسان گروه بیوانفورماتیک وانیار بررسی شده است.




عضویت در مجله وانیار
چطور از جدیدترین آموزشها باخبر شوم؟
با عضویت در مجله بیوانفورماتیک وانیار، برترین آموزشهای بیوانفورماتیک را در لحظه انتشار دریافت کنید.
سلام، وقت بخیر.
چطور میتونیم بهتون کمک کنیم؟
تیم ما آماده پاسخگویی به سوالات شماست.
پشتیبانی 24 ساعته در 7 روز هفته.